The Determination of Cellular Proteins
The traditional method of identifying a protein is to remove its amino acids one at a time and determine the identity of each one. This method is far too slow and labor-
To analyze a protein with mass spectrometry, researchers break it into small peptide fragments using a protein-
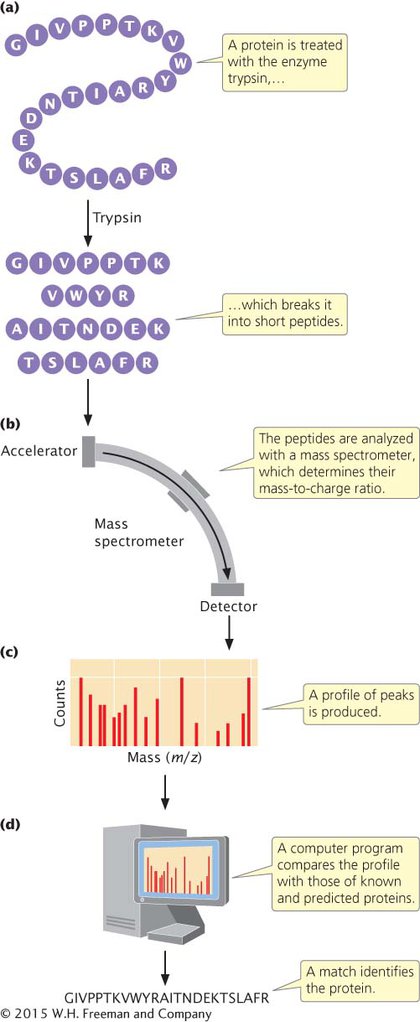
Mass spectrometric methods can also be used to measure the amount of each protein identified. In this procedure, a complex mixture of proteins (such as those from a tissue sample) is digested and analyzed with mass spectrometry. The computer program then sorts out the proteins present in the sample by analyzing the peptide profiles.
Mary Lipton and her colleagues used this approach to study the proteome of Deinococcus radiodurans, an exceptional bacterium that is able to withstand high doses of ionizing radiation that are lethal to all other organisms. The genome of D. radiodurans had already been sequenced. Lipton and her colleagues extracted proteins from the bacterium, broke them up into small peptide fragments, separated the fragments, and then used mass spectrometry to determine the proteins from the peptide fragments. They were able to identify 1910 proteins, which represented more than 60% of the proteins predicted on the basis of the genome sequence.