Chapter 30
(a) Pyruvate; (b) oxaloacetate; (c) α-ketoglutarate; (d) β-ketoisocaproate; (e) phenylpyruvate; (f) hydroxyphenylpyruvate
 Page C32
Page C32The required coenzymes are pyridoxal phosphate in the transamination reaction and NAD+/NADH in the redox reactions.
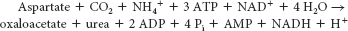
Most enzymes are specific for one or the other. Enzymes in catabolic pathways use NADH/NAD+, whereas enzymes in anabolic pathways use only NADPH/NADP+.
Aminotransferases transfer the α-amino group to α-ketoglutarate to form glutamate. Glutamate is oxidatively deaminated to form an ammonium ion.
Aspartate (oxaloacetate), glutamate (α-ketoglutarate), alanine (pyruvate)
Serine and threonine
Carbamoyl phosphate and aspartate
Complete the interactive matching exercise to see answers.

The number of high-
transfer- potential phosphoryl groups used remains four. The synthesis of fumarate by the urea cycle is important because it links the urea cycle and the citric acid cycle. Fumarate is hydrated to malate, which, in turn, is oxidized to oxaloacetate. Oxaloacetate has several possible fates: (1) transamination to aspartate, (2) conversion into glucose by the gluconeogenic pathway, (3) condensation with acetyl CoA to form citrate, or (4) conversion into pyruvate.
The analytical results strongly suggest that three enzymes—
pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and the branched- chain α-ketoacid dehydrogenase— are deficient. In fact, the E3 component (Chapter 18) is common to all of these enzymes. The results suggest that E3 is missing or defective. This proposal could be tested by purifying these three enzymes and assaying their ability to catalyze the regeneration of lipoamide. A, arginine; B, citrulline; C, ornithine; D, arginosuccinate. The order of appearance: C, B, D, E.
Aspartame, a dipeptide ester, is hydrolyzed to l-aspartate and l-phenylalanine. High levels of phenylalanine are harmful in phenylketonurics.
N-Acetylglutamate is synthesized from acetyl CoA and glutamate. Once again, acetyl CoA serves as an activated acetyl donor. This reaction is catalyzed by N-acetylglutamate synthase.
The carbon skeletons of ketogenic amino acids can be converted into ketone bodies or fatty acids. Only leucine and lysine are purely ketogenic. Glucogenic amino acids are those whose carbon skeletons can be converted into glucose.
Pyruvate (glycolysis and gluconeogenesis), acetyl CoA (citric acid cycle and fatty acid synthesis), acetoacetyl CoA (ketone-
body formation), α-ketoglutarate (citric acid cycle), succinyl CoA (citric acid cycle), fumarate (citric acid cycle), and oxaloacetate (citric acid cycle and gluconeogenesis) As shown in Figure 30.12, alanine, a gluconeogenic amino acid, is released during the metabolism of tryptophan to acetyl CoA and acetoacetyl CoA.
Not all proteins are created equal; some are more important than others. Some proteins would be degraded to provide the missing amino acid. The nitrogen from the other proteins would be excreted as urea. Consequently, more nitrogen would be excreted than ingested.
This defect can be partly bypassed by providing a surplus of arginine in the diet and restricting the total protein intake. In the liver, arginine is split into urea and ornithine, which then reacts with carbamoyl phosphate to form citrulline. This urea-
cycle intermediate condenses with aspartate to yield argininosuccinate, which is then excreted. Note that two nitrogen atoms— one from carbamoyl phosphate and the other from aspartate— are eliminated from the body per molecule of arginine provided in the diet. In essence, argininosuccinate substitutes for urea in carrying nitrogen out of the body. The formation of argininosuccinate removes the nitrogen, and the restriction on protein intake relieves the aciduria. Double-
displacement. A substituted enzyme intermediate is formed. The branched-
chain amino acids leucine, isoleucine, and valine. The required enzyme is the branched- chain α-ketoacid dehydrogenase complex. Ammonia could lead to the amination of α-ketoglutarate, producing a high concentration of glutamate in an unregulated fashion. α-Ketoglutarate for glutamate synthesis could be removed from the citric acid cycle, thereby diminishing the cell’s respiration capacity.
The liver is the primary tissue for capturing nitrogen as urea. If the liver is damaged (for instance, by hepatitis or the excessive consumption of alcohol), free ammonia is released into the blood.
Ornithine transcarbamoylase
Depletion of glycogen stores. When they are gone, proteins must be degraded to meet the glucose needs of the brain. The resulting amino acids are deaminated, and the nitrogen atoms are excreted as urea.
The brain has adapted to the use of ketone bodies, which are derived from fatty acid catabolism. In other words, the brain is being powered by fatty acid breakdown.
When the glycogen and lipid stores are gone, the only available energy source is protein.
Deamination to α-keto-
β-methylvalerate; oxidative decarboxylation to α-methylbutyryl CoA; oxidation to tiglyl CoA; hydration, oxidation, and thiolysis yields acetyl CoA and propionyl CoA; propionyl CoA to succinyl CoA. In the Cori cycle, the carbon atoms are transferred from muscle to the liver as lactate. For lactate to be of any use, it must be reduced to pyruvate. This reduction requires high-
energy electrons in the form of NADH. When the carbon atoms are transferred as alanine, transamination yields pyruvate directly. The precise cause of all of the symptoms is not firmly established, but a likely explanation depends on the centrality of oxaloacetate to metabolism. A lack of pyruvate carboxylase would reduce the amount of oxaloacetate. The lack of oxaloacetate would reduce the activity of the citric acid cycle, and so ATP would be generated by lactic acid formation. If the concentration of oxaloacetate is low, aspartate cannot be formed and the urea cycle would be compromised. Oxaloacetate is also required to form citrate, which transports acetyl CoA to the cytoplasm for fatty acid synthesis. Finally, oxaloacetate is required for gluconeogenesis.