Chapter 4
The energy barrier that must be crossed to go from the polymerized state to the hydrolyzed state is large even though the reaction is thermodynamically favorable.
(a) Alanine-
glycine- serine; (b) Alanine; (c and d): 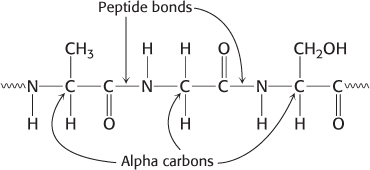
(a) TEPIVAPMEYGK; (b) −1 at pH 7; (c) −4 at pH 12
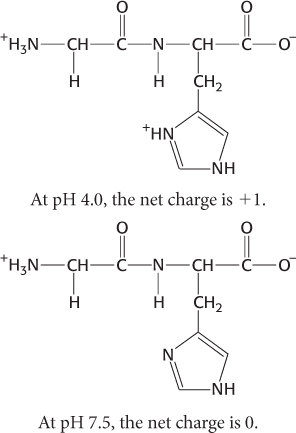
This observation demonstrates that pKa values are affected by the environment. A given amino acid can have a variety of pKa values, depending on the chemical environment inside the protein.
The peptide bond has partial double-
bond character, which prevents rotation. This lack of rotation constrains the conformation of the peptide backbone and limits possible structures. Complete the interactive matching exercise to see answers.
The translation of an α helix is 1.5 Å/amino acid. Therefore, the axial length of the helix would be 1.5 Å/amino acid ×120 amino acids = 180 Å. In a fully extended polypeptide chain, the distance between amino acids is 3.5 Å, so the length of the fully extended chain is 420 Å.
There are 20 choices for each of the 50 amino acids: 2050, or 1.13 × 1065, a very large number.
The (nitrogen–
α carbon– carbonyl carbon) repeating unit Side chain is the functional group attached to the α-carbon atom of an amino acid.
Amino acid composition refers simply to the amino acids that make up the protein. The order is not specified. Amino acid sequence is the same as the primary structure—
the sequence of amino acids from the amino terminal to the carboxyl terminal of the protein. Different proteins may have the same amino acid composition, but amino acid sequence identifies a unique protein. The primary structure determines the tertiary structure. Knowing the primary structure helps to elucidate the function of the protein. Knowledge of the primary structure of mutated proteins enables an understanding of the biochemical basis of some diseases. Primary structure can reveal the evolutionary history of the protein.
The helix is a condensed, coiled structure, with the R groups bristling outward from the axis of the helix. The distance between two adjacent amino acids is 1.5 Å. The strand is a fully extended polypeptide chain, and the side chains of adjacent amino acids point in opposite directions. The distance between adjacent amino acids is 4.5 Å. Both structures are stabilized by hydrogen bonding between components of the polypeptide backbone.
Primary structure—
peptide bond; secondary structure— local hydrogen bonds between components of the polypeptide backbone; tertiary structure— various types of noncovalent bonds between R groups that are far apart in the primary structure; quaternary structure— various noncovalent bonds between R groups on the surface of subunits. Complete the interactive matching exercise to see answers.
No, the Pro–
X bond would have the characteristics of any other peptide bond. The steric hindrance in X– Pro arises because the R group of Pro is bonded to the amino group. Hence, in X– Pro, the proline R group is near the R group of X, which would not be the case in Pro– X. The methyl group attached to the β-carbon atom of isoleucine sterically interferes with α-helix formation. In leucine, this methyl group is attached to the γ-carbon atom, which is farther from the main chain and hence does not interfere.
The first mutation destroys activity because valine occupies more space than alanine does, and so the protein must take a different shape, assuming that this residue lies in the closely packed interior. The second mutation restores activity because of a compensatory reduction of volume; glycine is smaller than isoleucine.
Page C4Loops invariably are on the surface of proteins, exposed to the environment. Because many proteins exist in aqueous environments, the exposed loops are hydrophilic so as to interact with water.
Heat would increase the thermal energy of the chain. The weak bonds holding the chain in its correct three-
dimensional structure would not be able to withstand the wiggling of the backbone, and the tertiary structure would be lost. Often, the denatured chains would interact with each other, forming large complexes that precipitate out of solution. Detergents would denature the protein by essentially turning it inside out. The hydrophobic residues in the interior of the protein would interact with the detergent, whereas the hydrophilic residues would interact with one another and not with the environment.
All ionic interaction, including hydrogen bonds, would be disrupted, resulting in protein denaturation.
Glycine has the smallest side chain of any amino acid. Its size is often critical in allowing polypeptide chains to make tight turns or to approach one another closely.
Glutamate, aspartate, and the terminal carboxylate can form salt bridges with the guanidinium group of arginine. In addition, this group can be a hydrogen-
bond donor to the side chains of glutamine, asparagine, serine, threonine, aspartate, glutamate, and tyrosine and to the main- chain carbonyl group. At pH 7, histidine also can hydrogen bond with arginine. Disulfide bonds in hair are broken by adding a thiol-
containing reagent and applying gentle heat. The hair is curled, and an oxidizing agent is added to reform disulfide bonds to stabilize the desired shape. Some proteins that span biological membranes are “the exceptions that prove the rule” because they have the reverse distribution of hydrophobic and hydrophilic amino acids. For example, consider porins, proteins found in the outer membranes of many bacteria. Membranes are built largely of hydrophobic chains. Thus, porins are covered on the outside largely with hydrophobic residues that interact with the neighboring hydrophobic chains. In contrast, the center of the protein contains many charged and polar amino acids that surround a water-
filled channel running through the middle of the protein. Thus, because porins function in hydrophobic environments, they are “inside out” relative to proteins that function in aqueous solution. The amino acids will be hydrophobic in nature. An α helix is especially suitable for crossing a membrane because all of the amide hydrogen atoms and carbonyl oxygen atoms of the peptide backbone take part in intrachain hydrogen bonds, thus stabilizing these polar atoms in a hydrophobic environment.
Recall that hemoglobin exists as a tetramer, whereas myoglobin is a monomer. Consequently, the hydrophobic residues on the surface of hemoglobin subunits probably take part in van der Waals interactions with similar regions of the other subunits and are shielded from the aqueous environment by these interactions.
A possible explanation is that the severity of the symptoms corresponds to the degree of structural disruption. Hence, the substitution of alanine for glycine might result in mild symptoms, but the substitution of the much larger tryptophan might prevent little or no collagen triple-
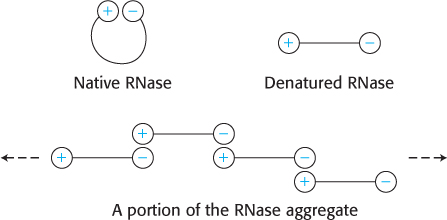
helix formation. The reason is that the wrong disulfides formed pairs in urea. There are 105 different ways of pairing eight cysteine molecules to form four disulfides; only one of these combinations is enzymatically active. The 104 wrong pairings have been picturesquely termed “scrambled” ribonuclease.
The added β-mercaptoethanol catalyzed the rearrangement of disulfide pairings until the native structure was regained. This process was driven by the decrease in free energy as the scrambled conformations were converted into the stable, native conformation of the enzyme. The native disulfide pairings of ribonuclease thus contribute to the stabilization of the thermodynamically preferred structure.
The native conformation of insulin is not the thermodynamically most stable form, because it contains two separate chains linked by disulfide bonds. Insulin is formed from proinsulin, a single-
chain precursor, which is cleaved to form insulin, a 51- residue molecule, after the disulfide bonds have formed. A segment of the main chain of the protease could hydrogen bond to the main chain of the target protein to form an extended parallel or antiparallel pair of β strands.
As the size of the protein increases, the surface-
to- volume ratio decreases. Consequently, the ratio of hydrophilic amino acids to hydrophobic amino acids also decreases. Each strand is 35 kDa and hence has about 318 residues (the mean residue mass is 110 Da). Because the rise per residue in an a helix is 1.5 Å, the length is 477 Å. More precisely, for an α-helical coiled coil, the rise per residue is 1.46 Å; so the length is 464 Å.
Most likely, the RNase molecules would have become tangled with one another to form a large, insoluble aggregate. For example, suppose a positively charged R group interacted with a negatively charged R group in the native structure. If the protein concentration were high upon denaturation, groups from different molecules might interact to form a large, insoluble aggregate.