
13.2 Receptor Proteins Transmit Information into the Cell
✓ 5 Differentiate the various types of membrane receptors.
Most receptor proteins that convey environmental information into the cell interior fall into three classes: seven-
Seven-Transmembrane-Helix Receptors Change Conformation in Response to Ligand Binding and Activate G Proteins

The seven-


 Figure 13.3 The structure of 7TM receptors. (A) Schematic representation of a 7TM receptor showing how it passes through the membrane seven times. (B) Three-
Figure 13.3 The structure of 7TM receptors. (A) Schematic representation of a 7TM receptor showing how it passes through the membrane seven times. (B) Three-The 7TM receptors undergo a change in conformation in response to ligand binding. The binding of a ligand on the outside the cell induces a conformational change in the 7TM receptor that can be detected inside the cell. As we shall see, 7TM receptors also have in common the next step in their signal-
Ligand Binding to 7TM Receptors Leads to the Activation of G Proteins
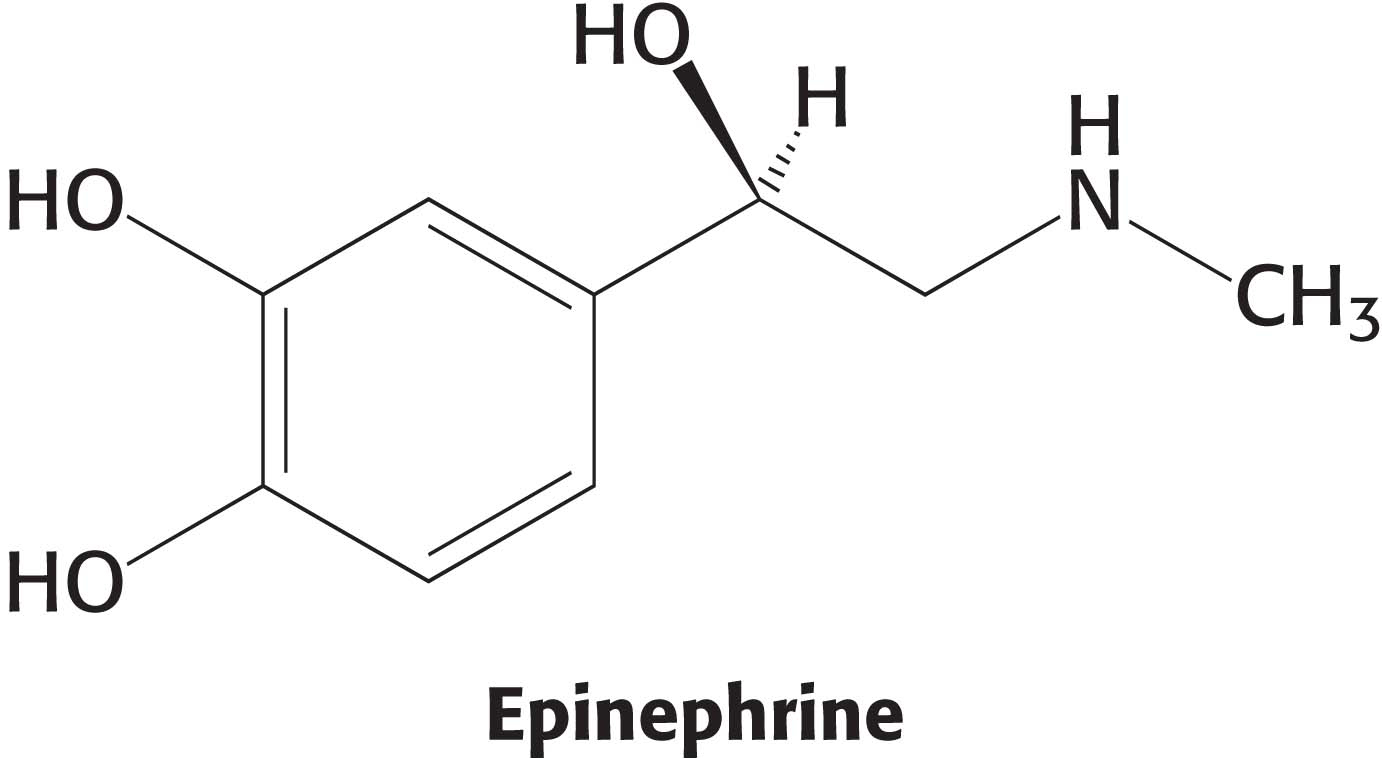
Let us focus on the β-adrenergic receptor as a model of the 7TM receptor class. What is the next step in the pathway after the binding of epinephrine by the β-adrenergic receptor? The conformational change in the cytoplasmic domain of the receptor activates a GTP-

How do these G proteins operate? In the unactivated state, the guanyl nucleotide bound to the G protein is GDP. In this form, the G protein exists as a heterotrimer consisting of α, β, and γ subunits; the a subunit (referred to as Gα) binds the nucleotide (Figure 13.5). The α and γ subunits are usually anchored to the membrane by covalently attached fatty acids. The exchange of the bound GDP for GTP is catalyzed by the ligand-
 A heterotrimeric G protein. (A) A ribbon diagram shows the relation between the three subunits. In this complex, the α subunit (gray and purple) is bound to GDP. Notice that GDP is bound in a pocket close to the surface at which the α subunit interacts with the βγ dimer (the β subunit is shown in blue and γ in yellow). (B) A schematic representation of the heterotrimeric G protein.
A heterotrimeric G protein. (A) A ribbon diagram shows the relation between the three subunits. In this complex, the α subunit (gray and purple) is bound to GDP. Notice that GDP is bound in a pocket close to the surface at which the α subunit interacts with the βγ dimer (the β subunit is shown in blue and γ in yellow). (B) A schematic representation of the heterotrimeric G protein.
A single ligand–
Activated G Proteins Transmit Signals by Binding to Other Proteins
As just described, the formation of the ligand–
 Adenylate cyclase activation. (A) Adenylate cyclase is a membrane protein with two large intracellular domains (red and orange) that contain the catalytic apparatus. (B) The structure of the complex between Gα in its GTP form bound to a catalytic fragment of adenylate cyclase. Notice that the surface of Gα that had been bound to the bg dimer (Figure 13.5) now binds adenylate cyclase.
Adenylate cyclase activation. (A) Adenylate cyclase is a membrane protein with two large intracellular domains (red and orange) that contain the catalytic apparatus. (B) The structure of the complex between Gα in its GTP form bound to a catalytic fragment of adenylate cyclase. Notice that the surface of Gα that had been bound to the bg dimer (Figure 13.5) now binds adenylate cyclase.
Cyclic AMP Stimulates the Phosphorylation of Many Target Proteins by Activating Protein Kinase A
The increased concentration of cAMP can affect a wide range of cellular processes, depending on the cell type. For example, it enhances the degradation of storage fuels, increases the secretion of acid by the gastric mucosa in the cells of the stomach and intestines, leads to the dispersion of melanin pigment granules in skin cells, diminishes the aggregation of blood platelets, and induces the opening of chloride channels in the pancreas. How does cAMP affect so many cellular processes? Is there a common denominator for its diverse effects? Indeed there is. Most effects of cAMP in eukaryotic cells are mediated by the activation of a single protein kinase. This key enzyme is called protein kinase A (PKA). Kinases are enzymes that phosphorylate a substrate at the expense of a molecule of ATP. PKA consists of two regulatory (R) subunits and two catalytic (C) subunits. In the absence of cAMP, the R2C2 complex is catalytically inactive (Figure 13.7). The binding of cAMP to the regulatory subunits releases the catalytic subunits, which are enzymatically active on their own. Activated PKA then phosphorylates specific serine and threonine residues in many targets to alter their activity. The alteration in activity is due to structural and ionic changes that result from the introduction of the large negatively charged phosphate functional group. The cAMP cascade is turned off by cAMP phosphodiesterase, an enzyme that converts cAMP into AMP, which does not activate PKA. The C and R subunits subsequently rejoin to form the inactive enzyme.

 CLINICAL INSIGHT
CLINICAL INSIGHTMutations in Protein Kinase A Can Cause Cushing’s Syndrome
Cushing’s syndrome, a collection of diseases resulting from excess cortisol secretion by the adrenal cortex, is a metabolic disorder characterized by a variety of symptoms such as muscle weakness, thinning skin that is easily bruised, and osteoporosis. Cortisol, a steroid hormone (Section 29.5), has a number of physiological effects including stimulation of glucose synthesis, suppression the immune response, and inhibition of bone growth. The most common cause of Cushing’s syndrome, called Cushing’s disease, is a tumor of the pituitary gland that overstimulates cortisol secretion by the adrenal cortex. Recent work shows that a mutation that renders protein kinase A constitutively active also results in the syndrome. In these patients, the catalytic subunit of the enzyme is altered so that it no longer binds the regulatory subunit. Thus, the enzyme is active even in the absence of cAMP with the ultimate result being unregulated secretion of cortisol.
G Proteins Spontaneously Reset Themselves Through GTP Hydrolysis
The ability to shut down signal-

The ligand-
QUICK QUIZ 1
List the means by which the β-adrenergic pathway is terminated.
Dissociation of epinephrine from the receptor. Conversion of cAMP into AMP by phosphodiesterase and the subsequent inhibition of PKA. Conversion of GTP into GDP by Gα and the subsequent reformation of the inactive heterotrimeric G protein.

 CLINICAL INSIGHT
CLINICAL INSIGHTCholera and Whooping Cough Are Due to Altered G-Protein Activity
The alteration of G-
Whereas cholera is a result of a G protein trapped in the active conformation, causing the signal-
The Hydrolysis of Phosphatidylinositol Bisphosphate by Phospholipase C Generates Two Second Messengers
Cyclic AMP is not the only second messenger employed by 7TM receptors and the G proteins. We turn now to another ubiquitous second-

What are the biochemical effects of the second messenger IP3? Unlike cAMP, IP3 does not cause a cascade of phosphorylation to elicit a response from the cell. IP3 directly causes the rapid release of Ca2+ from intracellular stores—
The lifetime of IP3 in the cell is very short—
Diacylglycerol, the other molecule formed by the receptor-
