
29.4 Lipoproteins Transport Cholesterol and Triacylglycerols Throughout the Organism
Cholesterol and triacylglycerols are made primarily in the liver, but they are used by tissues throughout the body. How are these important but hydrophobic biochemicals shuttled through the bloodstream? Cholesterol and triacylglycerols are packaged into lipoprotein particles for transport through bodily fluids. Each particle consists of a core of hydrophobic lipids surrounded by a shell of more-
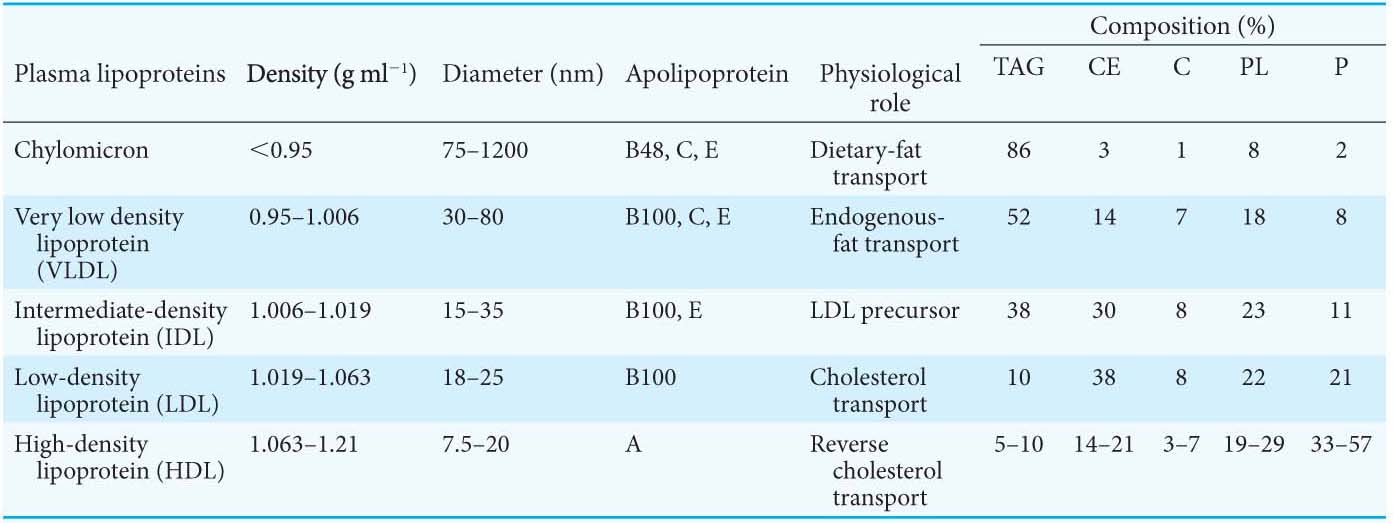
Lipoprotein particles are classified according to increasing density: chylomicrons, chylomicron remnants, very low density lipoproteins (VLDLs), intermediate-
Triacylglycerols and cholesterol in excess of the liver’s own needs are exported into the blood in the form of very low density lipoprotein. Triacylglycerols in VLDL are hydrolyzed by lipases on capillary surfaces, and the freed fatty acids are taken into the cells. The cholesterol-
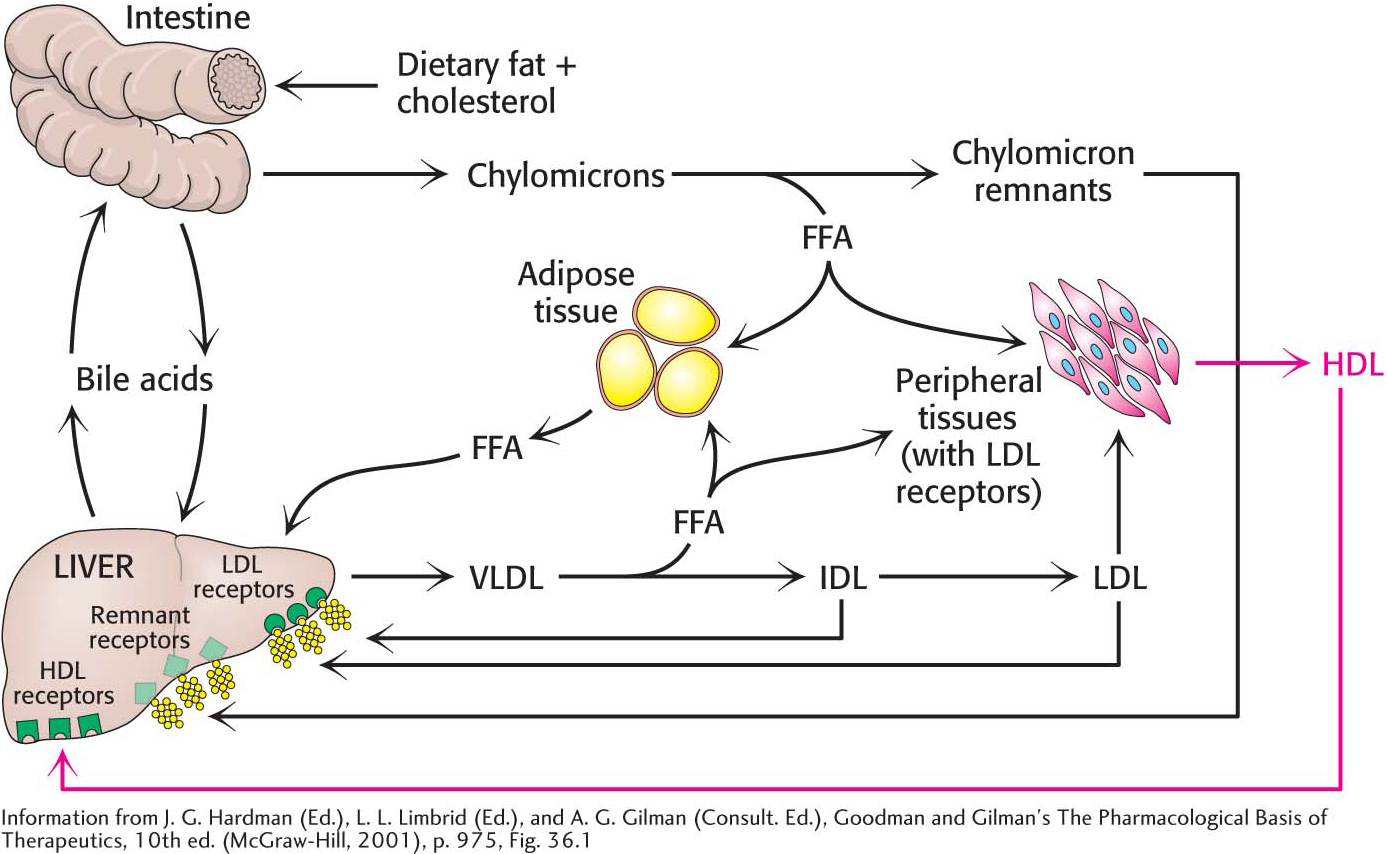
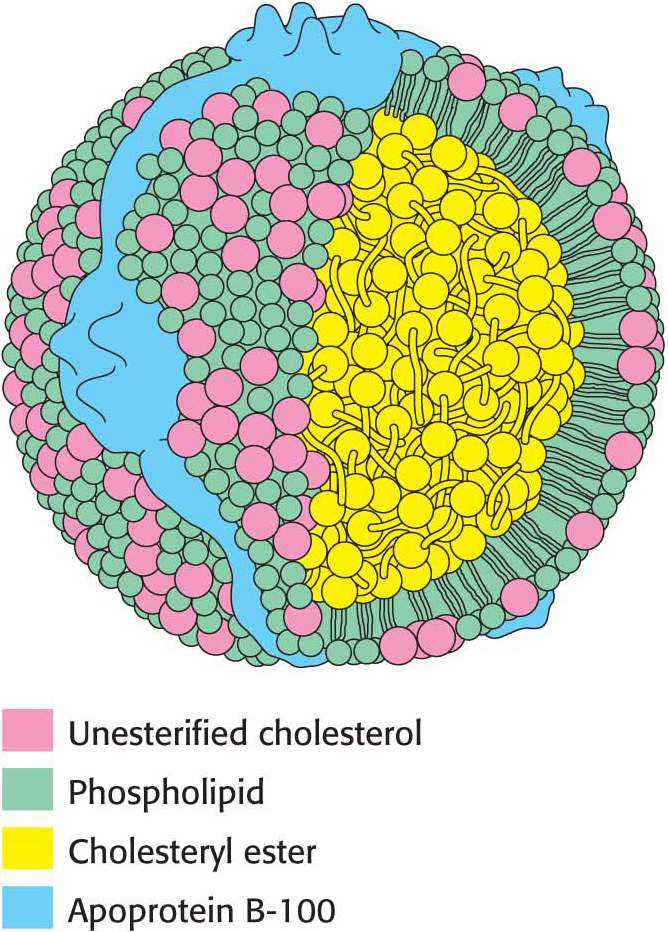
Low-
A different purpose is served by high-
Low-Density Lipoproteins Play a Central Role in Cholesterol Metabolism
Cholesterol metabolism must be precisely regulated to prevent atherosclerosis, the thickening of arterial walls with a subsequent loss of elasticity. The mode of control in the liver, the primary site of cholesterol synthesis, has already been discussed. The primary source of cholesterol for peripheral tissues is the low-
Receptor-
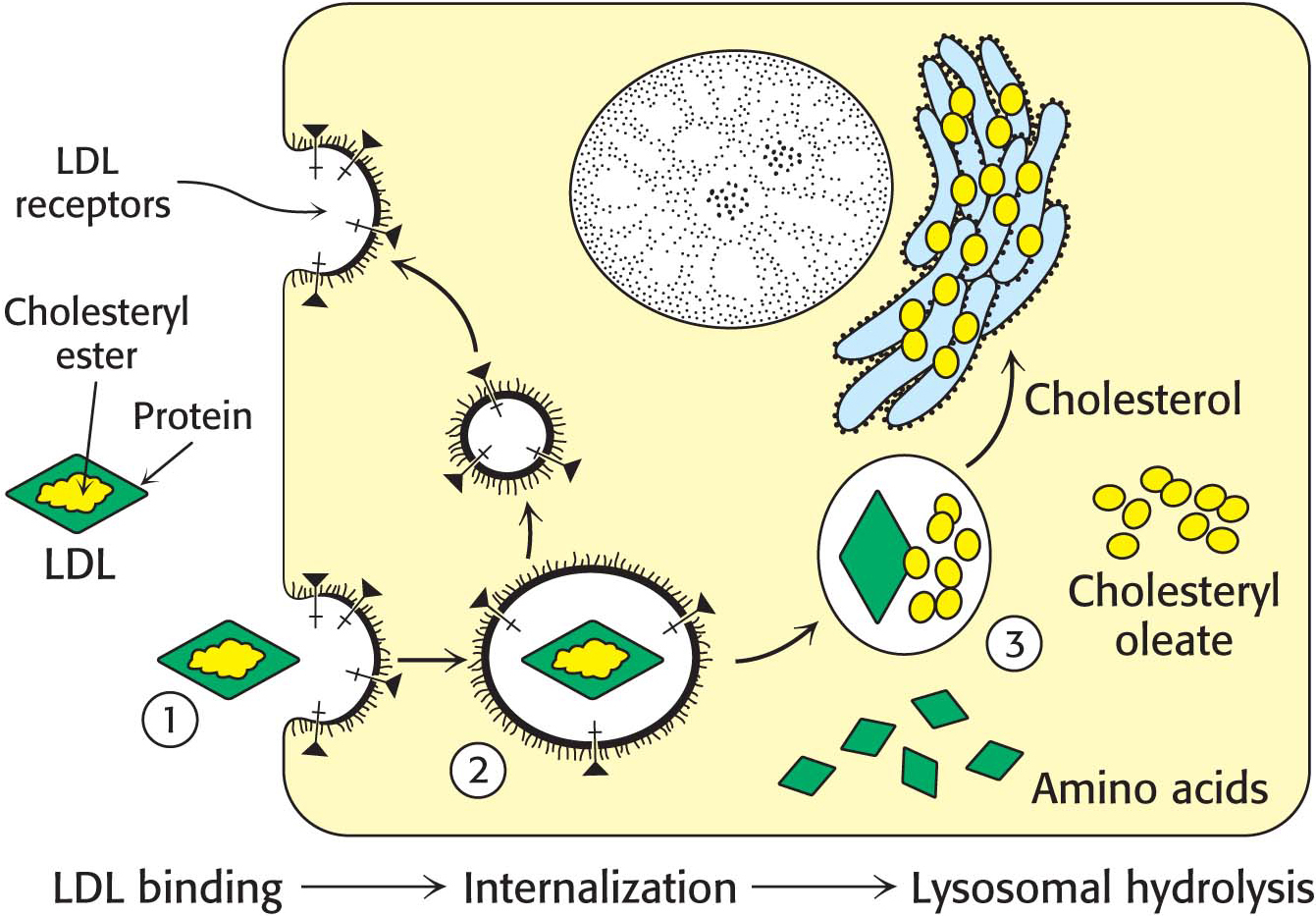
Low-
density lipoprotein binds to a receptor protein on the cell surface . Apoprotein B-100 on the surface of an LDL particle binds to a specific receptor protein on the plasma membrane of nonliver cells. The receptors for LDL are localized in specialized regions called coated pits, surrounded by a specialized protein called clathrin. Page 536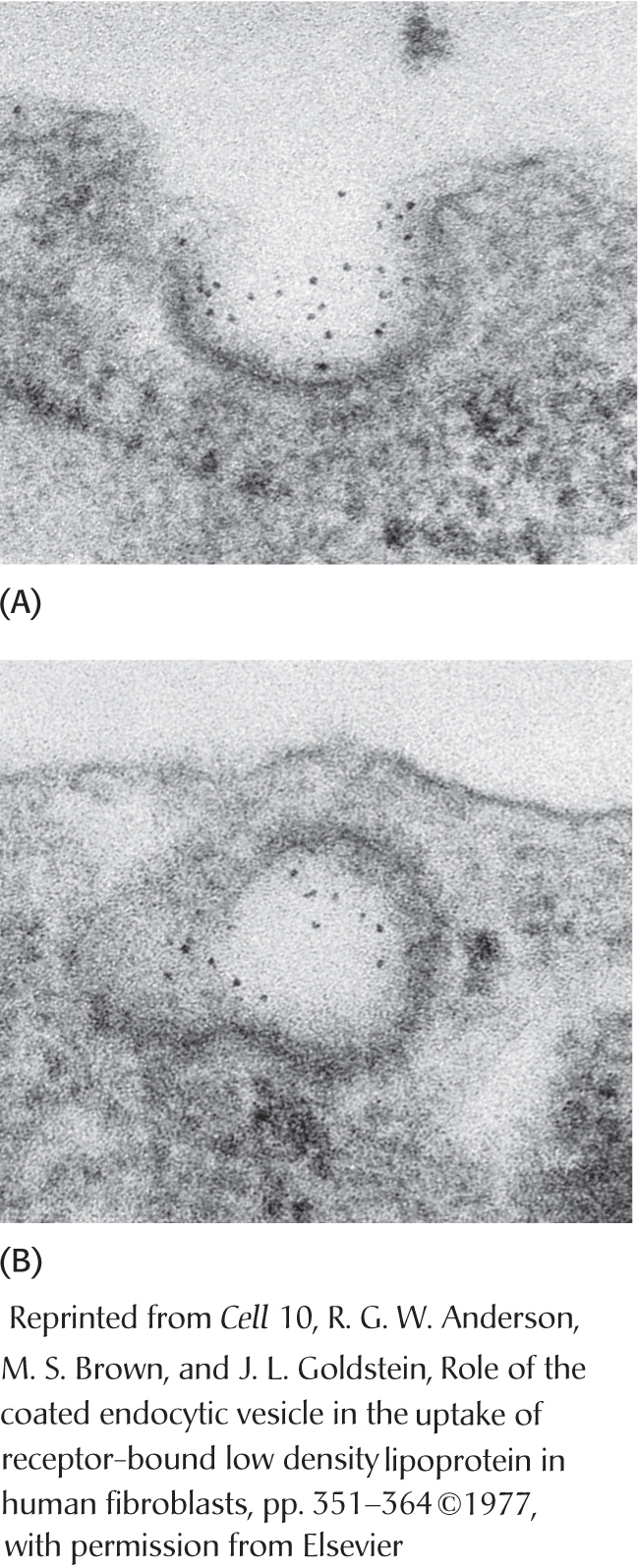 Figure 29.16 Endocytosis of low-
Figure 29.16 Endocytosis of low-density lipoprotein bound to its receptor . Micrographs of (A) LDL (conjugated to iron-laden ferritin for visualization, dark spots) bound to a coated- pit region on the surface of a cultured human fibroblast cell and (B) this region invaginating and fusing to form an endocytic vesicle. The cell internalizes the receptor–
LDL complex . The plasma membrane in the vicinity of the complex folds in on itself (invaginates). The membrane then fuses to form an endocytic vesicle (called an endosome), enclosing the receptor–LDL complex (endocytosis) (Figure 29.16). Low-
density lipoprotein is hydrolyzed in lysosomes . The vesicles containing LDL subsequently fuse with lysosomes, acidic vesicles that carry a wide array of degradative enzymes. The protein component of the LDL is hydrolyzed to free amino acids. The cholesteryl esters in the LDL are hydrolyzed by a lysosomal acid lipase. The LDL receptor itself usually returns unscathed to the plasma membrane. The round-trip time for a receptor is about 10 minutes; in its lifetime of about a day, it brings many LDL particles into the cell.
The released unesterified cholesterol can then be used for membrane biosynthesis or reesterified for storage inside the cell. The stored cholesterol must be reesterified because high concentrations of unesterified cholesterol disrupt the integrity of cell membranes.
The synthesis of the LDL receptor is itself subject to feedback regulation. Studies show that, when cholesterol is abundant inside the cell, new LDL receptors are not synthesized, blocking the uptake of additional cholesterol from plasma LDL.
 CLINICAL INSIGHT
CLINICAL INSIGHTThe Absence of the LDL Receptor Leads to Familial Hypercholesterolemia and Atherosclerosis
High cholesterol levels promote atherosclerosis, which is the leading cause of death in industrialized societies. Cholesterol’s role in the development of atherosclerosis was elucidated by the study of familial hypercholesterolemia, a genetic disorder. Familial hypercholesterolemia is characterized by high concentrations of cholesterol and LDL in the plasma, about three to four times the desired amount. In familial hypercholesterolemia, cholesterol is deposited in various tissues because of the high concentration of LDL cholesterol in the plasma. Nodules of cholesterol called xanthomas are prominent in skin and tendons in those having high levels of LDL. LDL also accumulates under the endothelial cells lining the blood vessels. Of particular concern is the oxidation of the excess LDL to form oxidized LDL (oxLDL), which can instigate the inflammatory response by the immune system, a response that has been implicated in the development of cardiovascular disease. The oxLDL is taken up by immune-
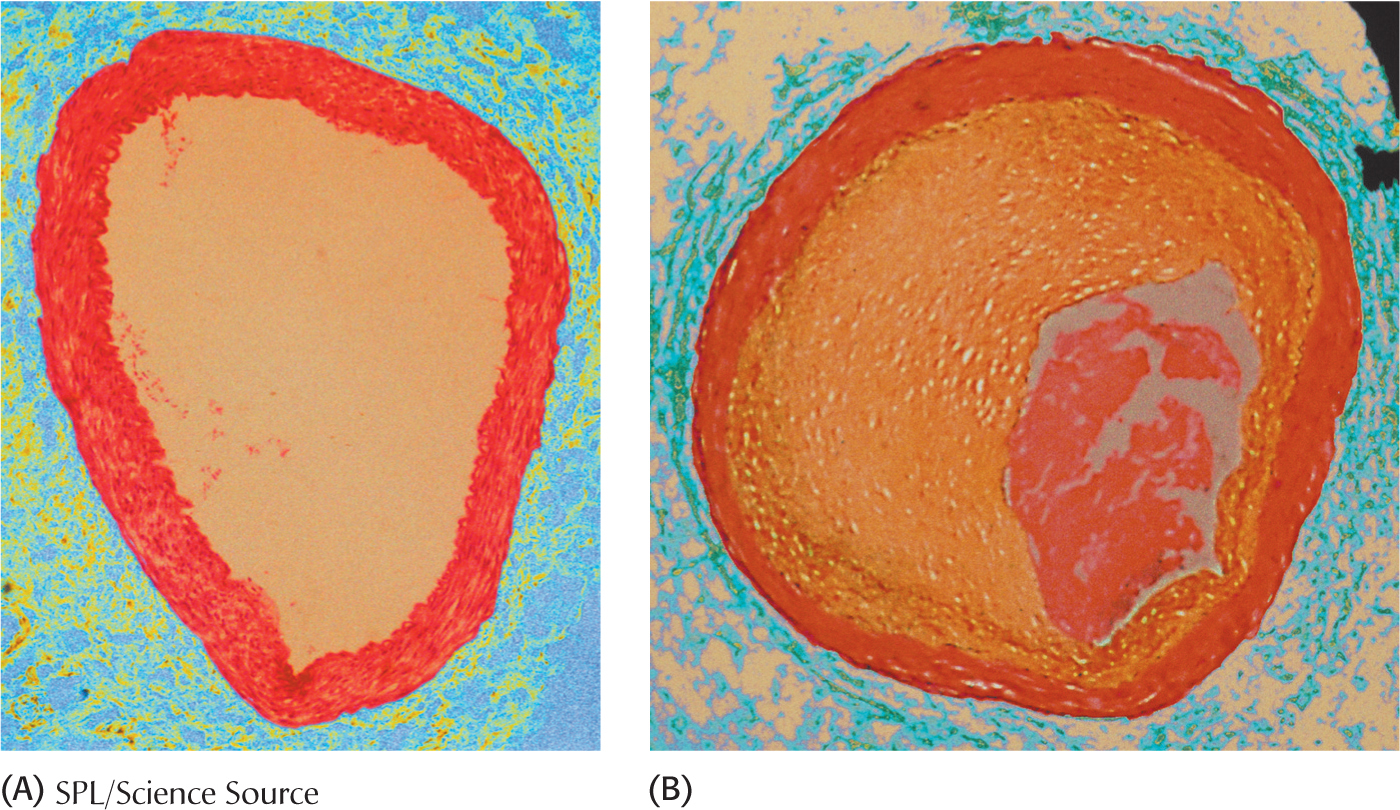
The molecular defect in most cases of familial hypercholesterolemia is an absence or deficiency of functional receptors for LDL. Homozygotes have almost no functional receptors for LDL, whereas heterozygotes have about half the normal number. Consequently, the entry of LDL into liver and other cells is impaired, leading to an increased plasma level of LDL. Most homozygotes die of coronary artery disease in childhood. The disease in heterozygotes (1 in 500 people) has a milder and more variable clinical course.
CLINICAL INSIGHTCycling of the LDL Receptor Is Regulated
PCSK9 (proprotein convertase subtilisin/kexin type 9) is a protease, secreted by the liver, that plays a crucial role in the regulation of cycling of the LDL receptor. Despite the fact that PCSK9 is a protease, enzymatic activity of the protein is not required for cycling regulation. PCSK9 in the blood binds to a domain on the receptor that prevents the receptor from returning to the plasma membrane, and it is degraded in the lysosome along with its cargo.
Individuals having a mutation that reduces the amount of PCSK9 in the blood have greatly reduced levels of LDL in the blood and display an almost 90% reduction in the rate of cardiovascular disease. Presumably, reduced levels of PCSK9 allow more receptor cycling and more efficient removal of LDL from the blood. Much research is now being directed at inhibiting PCSK9 activity in individuals with high cholesterol levels.
CLINICAL INSIGHTHDL Seems to Protect Against Atherosclerosis
Although the events that result in atherosclerosis take place rapidly in patients with familial hypercholesterolemia, a similar sequence of events take place in people who develop atherosclerosis over decades. In particular, the formation of foam cells and plaques are especially hazardous occurrences. HDL and its role in returning cholesterol to the liver are important in mitigating these life-
HDL has a number of antiatherogenic properties, including the inhibition of LDL oxidation, but the best-
The importance of reverse cholesterol transport is illustrated by the occurrence of mutations that inactivate a cholesterol-
Until recently, high levels of HDL-
CLINICAL INSIGHTThe Clinical Management of Cholesterol Levels Can Be Understood at a Biochemical Level
Homozygous familial hypercholesterolemia can be treated only by a liver transplant. A more generally applicable therapy is available for heterozygotes and others with high levels of cholesterol. The goal is to reduce the amount of cholesterol in the blood by stimulating the single normal gene to produce more than the customary number of LDL receptors. We have already observed that the production of LDL receptors is controlled by the cell’s need for cholesterol. Therefore, the strategy is to deprive the cell of ready sources of cholesterol. When cholesterol is required, the amount of mRNA for the LDL receptor rises and more receptor is found on the cell surface. This state can be induced by a two-
The reabsorption of bile is impeded by the oral administration of positively charged polymers, such as cholestyramine, that bind negatively charged bile salts and are not themselves absorbed. Cholesterol synthesis can be effectively blocked by a class of compounds called statins. A well-
