
30.3 Carbon Atoms of Degraded Amino Acids Emerge As Major Metabolic Intermediates
✓ 2 Explain how the carbon skeletons of the amino acids are metabolized after nitrogen removal.
We now turn to the fates of the carbon skeletons of amino acids after the removal of the α-amino group. The strategy of amino acid degradation is to transform the carbon skeletons into major metabolic intermediates that can be converted into glucose or oxidized by the citric acid cycle. The conversion pathways range from extremely simple to quite complex. The carbon skeletons of the diverse set of 20 fundamental amino acids are funneled into only seven molecules: pyruvate, acetyl CoA, acetoacetyl CoA, α-ketoglutarate, succinyl CoA, fumarate, and oxaloacetate. The conversion of 20 amino acid carbon skeletons into only seven molecules illustrates the remarkable economy of metabolic conversions, as well as the importance of certain metabolites.
Amino acids that are degraded to acetyl CoA or acetoacetyl CoA are termed ketogenic amino acids because they can give rise to ketone bodies or fatty acids but cannot be used to synthesize glucose. Recall that mammals lack a pathway for the net synthesis of glucose from acetyl CoA or acetoacetyl CoA. Amino acids that are degraded to pyruvate, α-ketoglutarate, succinyl CoA, fumarate, or oxaloacetate are termed glucogenic amino acids. Oxaloacetate, generated from pyruvate and other citric acid cycle intermediates, and pyruvate can be converted into phosphoenolpyruvate and then into glucose through gluconeogenesis.
Of the basic set of 20 amino acids, only leucine and lysine are solely ketogenic (Figure 30.5). Threonine, isoleucine, phenylalanine, tryptophan, and tyrosine are both ketogenic and glucogenic. Some of their carbon atoms emerge in acetyl CoA or acetoacetyl CoA, whereas others appear in potential precursors of glucose. The other 14 amino acids are classified as solely glucogenic. We will identify the degradation pathways by the entry point into metabolism. Additionally, because some degradation pathways are quite complex, they will be presented in outline form, with only some of the key intermediates highlighted.
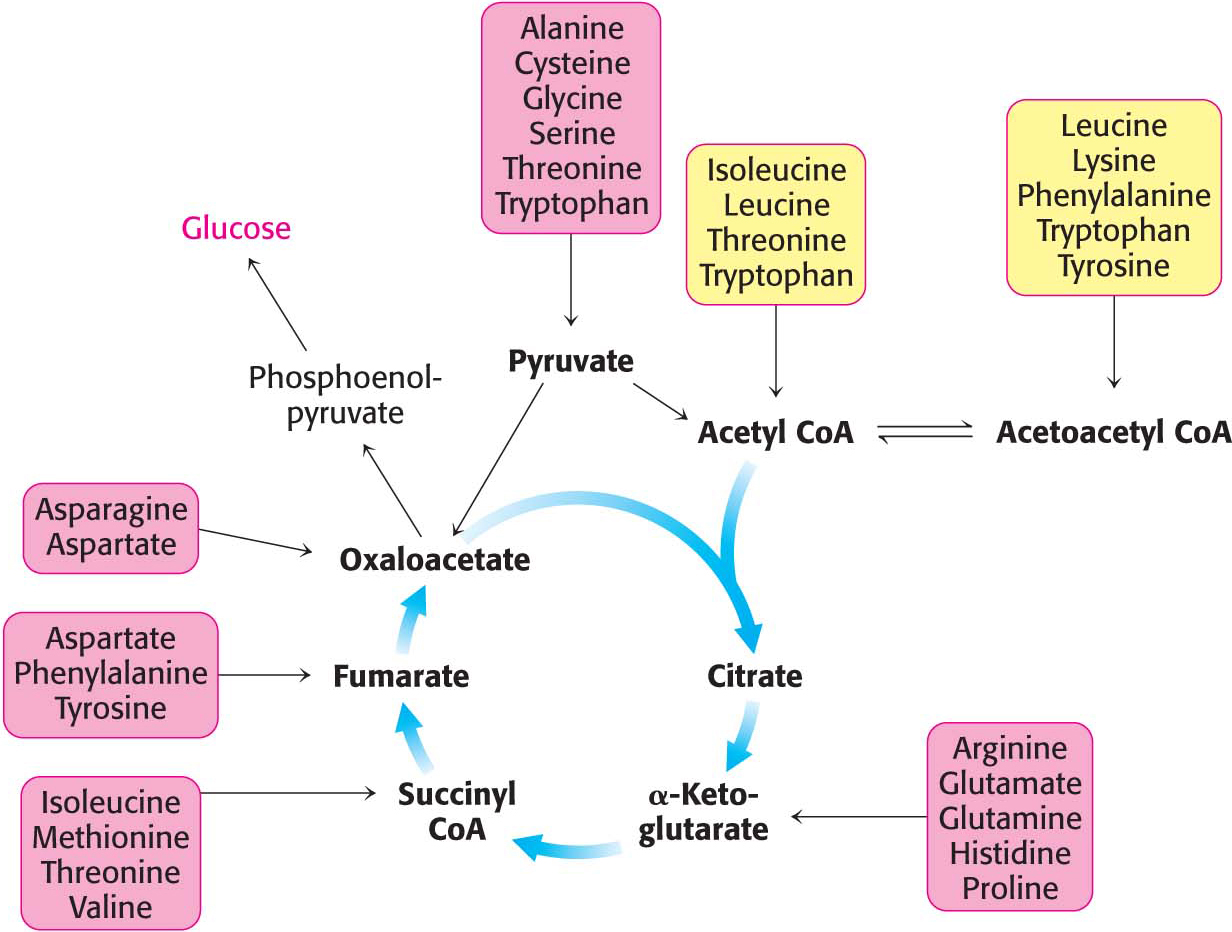
Pyruvate Is a Point of Entry into Metabolism
Pyruvate is the entry point of the three-
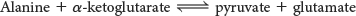
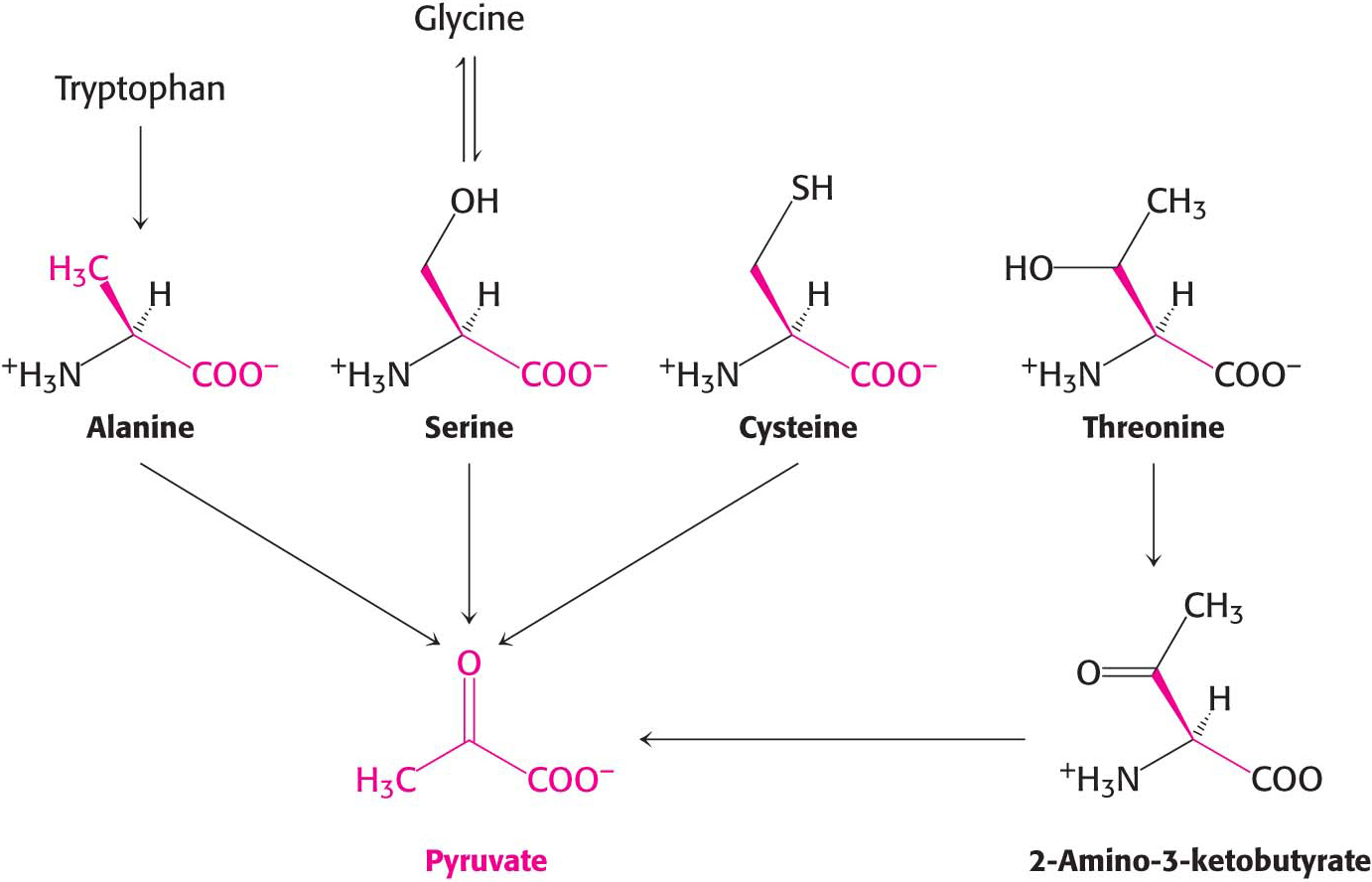
Another simple reaction in the degradation of amino acids is the deamination of serine to pyruvate by serine dehydratase:
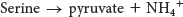
The conversion of cysteine into pyruvate is more complex. The conversion can take place through several pathways, with cysteine’s sulfur atom emerging in H2S, SCN–, or SO32–
The carbon atoms of three other amino acids also can be converted into pyruvate. Glycine can be converted into serine by the enzymatic addition of a hydroxymethyl group, or it can be cleaved to yield CO2, NH4+, and the activated one-
Oxaloacetate Is Another Point of Entry into Metabolism
Aspartate and asparagine are converted into oxaloacetate, a citric acid cycle intermediate. Aspartate, a four-

Asparagine is hydrolyzed by asparaginase to NH4+ and aspartate, which is then transaminated, as shown in the preceding reaction.
Recall that aspartate can also be converted into fumarate by the urea cycle. Fumarate is also a point of entry for half the carbon atoms of tyrosine and phenylalanine, as will be discussed shortly.
Alpha-Ketoglutarate Is Yet Another Point of Entry into Metabolism
The carbon skeletons of several five-
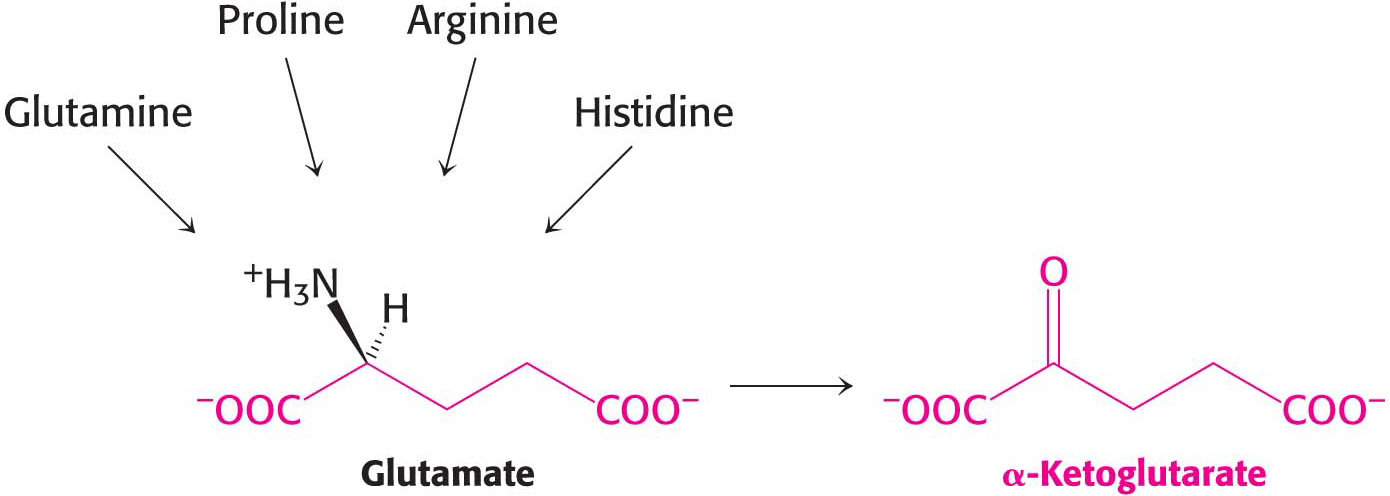
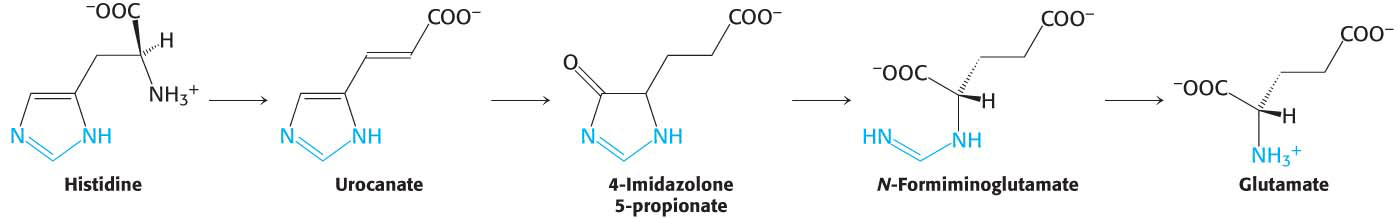
Histidine is converted into glutamate through the intermediate 4-
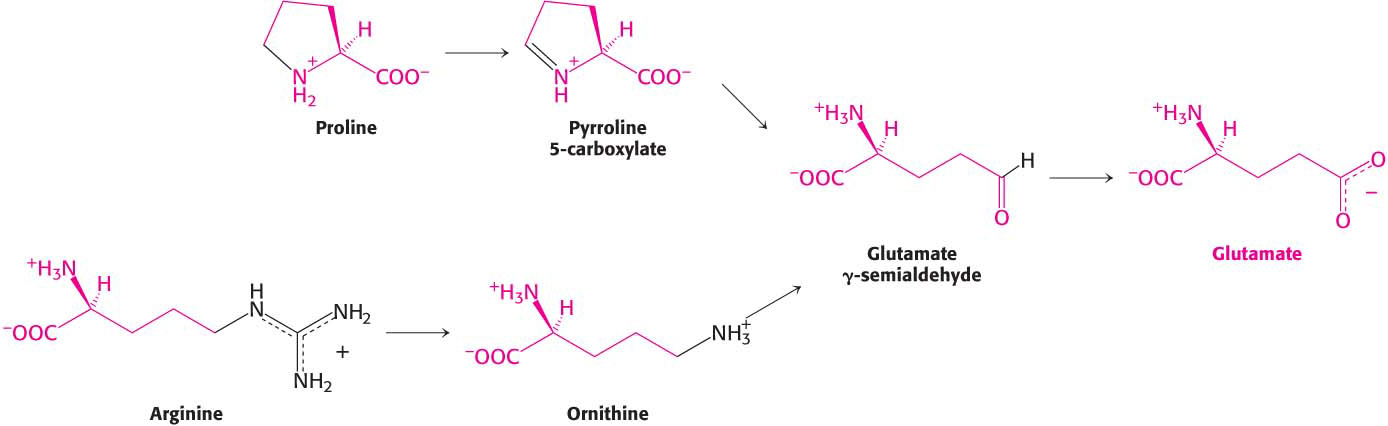
Glutamine is hydrolyzed to glutamate and NH4+ by glutaminase. Proline and arginine are each converted into glutamate γ-semialdehyde, which is then oxidized to glutamate (Figure 30.9).
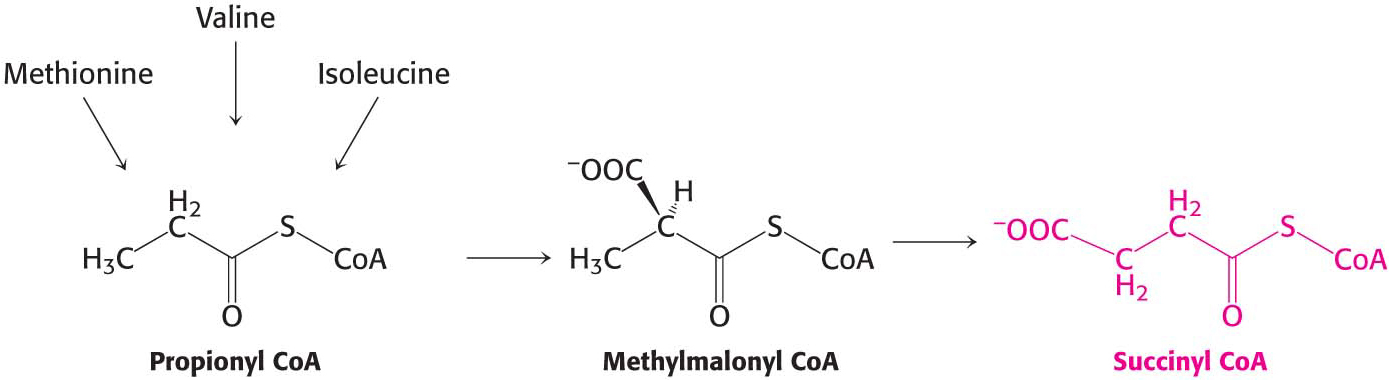
Succinyl Coenzyme A Is a Point of Entry for Several Nonpolar Amino Acids
Succinyl CoA is a point of entry for some of the carbon atoms of methionine, isoleucine, and valine. Propionyl CoA and then methylmalonyl CoA are intermediates in the breakdown of these three nonpolar amino acids (Figure 30.10). This pathway from propionyl CoA to succinyl CoA is also used in the oxidation of odd-
The Branched-Chain Amino Acids Yield Acetyl Coenzyme A, Acetoacetate, or Succinyl Coenzyme A
The degradation of the branched-
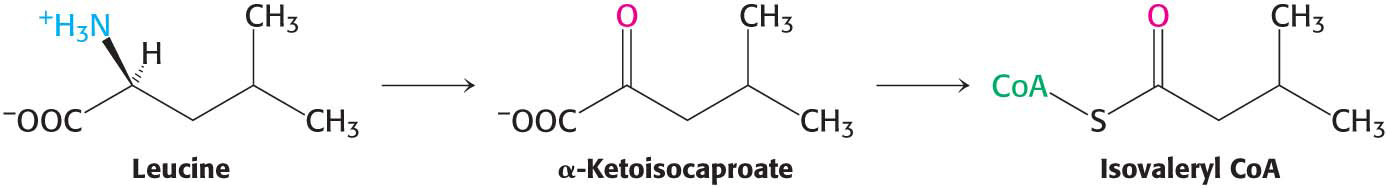
The isovaleryl CoA derived from leucine is dehydrogenated to yield β-methylcrotonyl CoA. The hydrogen acceptor is FAD, as in the analogous reaction in fatty acid oxidation. β-Methylglutaconyl CoA is then formed by the carboxylation of β-methylcrotonyl CoA at the expense of the hydrolysis of a molecule of ATP in a reaction similar to that of pyruvate carboxylase and acetyl CoA carboxylase:
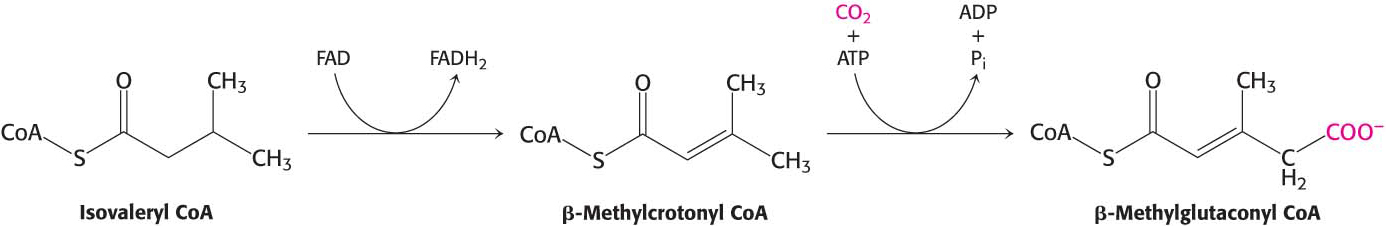
β-Methylglutaconyl CoA is hydrated to form 3-
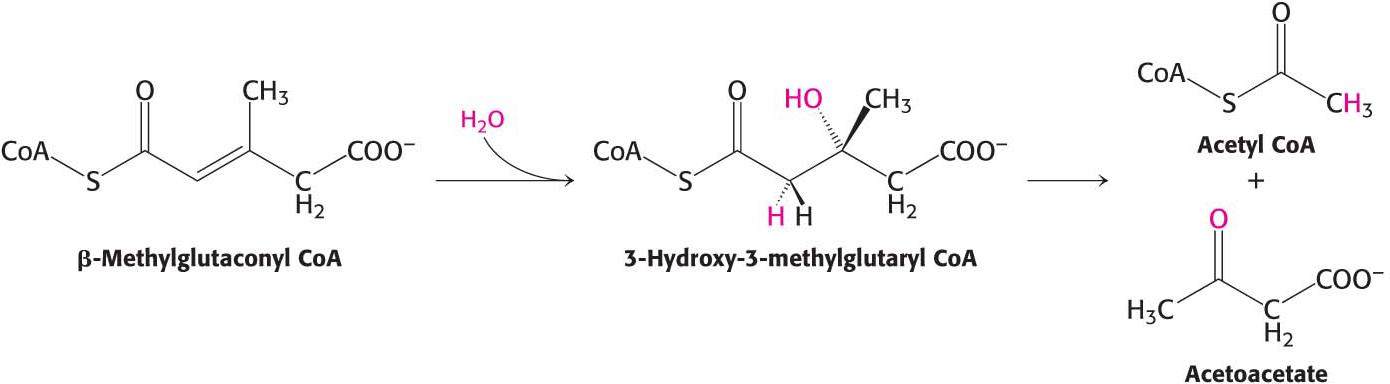
The degradative pathways of valine and isoleucine resemble that of leucine. After transamination and oxidative decarboxylation to yield a coenzyme A derivative, the subsequent reactions are like those of fatty acid oxidation. Isoleucine yields acetyl CoA and propionyl CoA, whereas valine yields CO2 and propionyl CoA. Propionyl CoA can be converted into the citric acid cycle intermediate succinyl CoA. The degradation of leucine, valine, and isoleucine confirm a point made earlier (Chapter 15): the number of reactions in metabolism is large, but the number of kinds of reactions is relatively small. The degradation of leucine, valine, and isoleucine provides a striking illustration of the underlying simplicity and elegance of metabolism.
Oxygenases Are Required for the Degradation of Aromatic Amino Acids
The degradation of the aromatic amino acids is not as straightforward as that of the amino acids considered so far, although the final products—
The degradation of phenylalanine begins with its hydroxylation to tyrosine, a reversible reaction catalyzed by phenylalanine hydroxylase. This enzyme is called a monooxygenase or mixed-
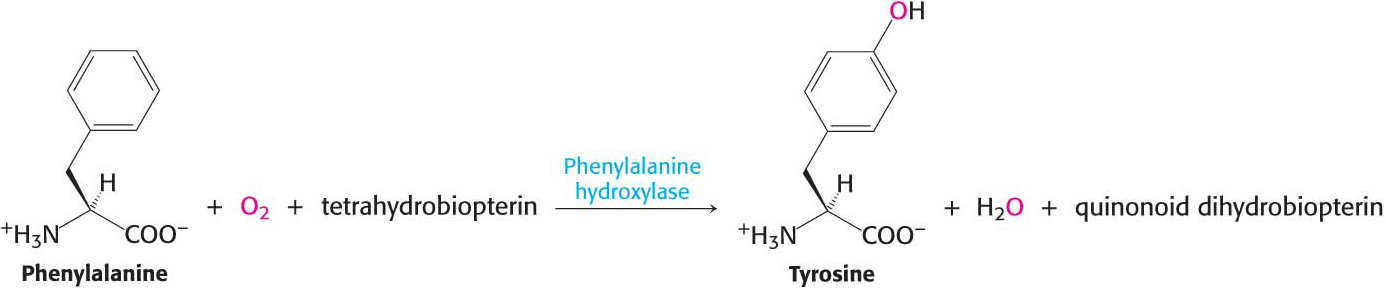
The reductant here is tetrahydrobiopterin, a cofactor of phenylalanine hydroxylase derived from the cofactor biopterin. NADH is used to regenerate tetrahydrobiopterin from the quinonoid form that was produced in the hydroxylation reaction. The sum of the reactions is
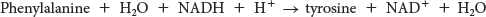
Note that these reactions can also be used to synthesize tyrosine from phenylalanine.
The next step in the degradation of phenylalanine and tyrosine is the transamination of tyrosine to p-
QUICK QUIZ 3
What are the common features of the breakdown products of the carbon skeletons of amino acids?
They are either fuels for the citric acid cycle, components of the citric acid cycle, or molecules that can be converted into a fuel for the citric acid cycle in one step.
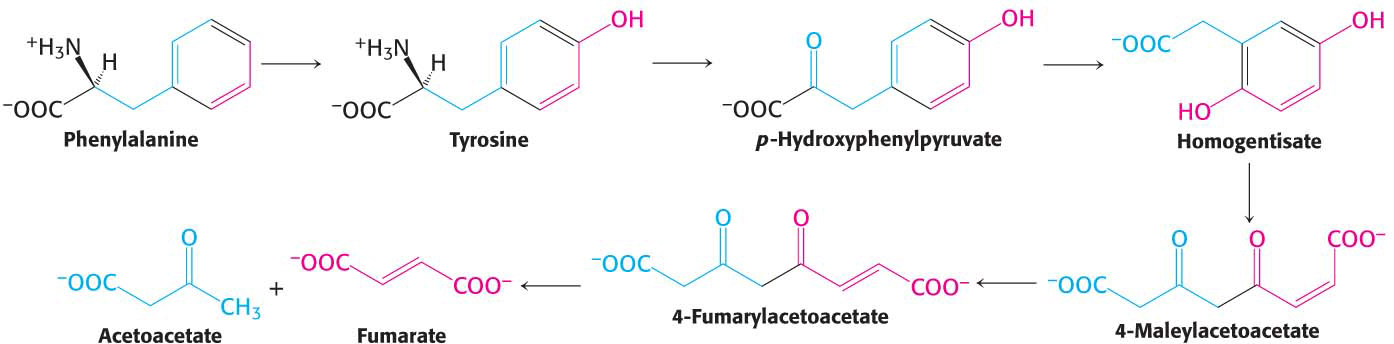
Tryptophan degradation requires several oxygenases to cleave its two rings to ultimately yield acetoacetate (Figure 30.12). Nearly all cleavages of aromatic rings in biological systems are catalyzed by dioxygenases. In contrast with monooxygenases, dioxygenases incorporate both oxygen atoms into the reaction product.
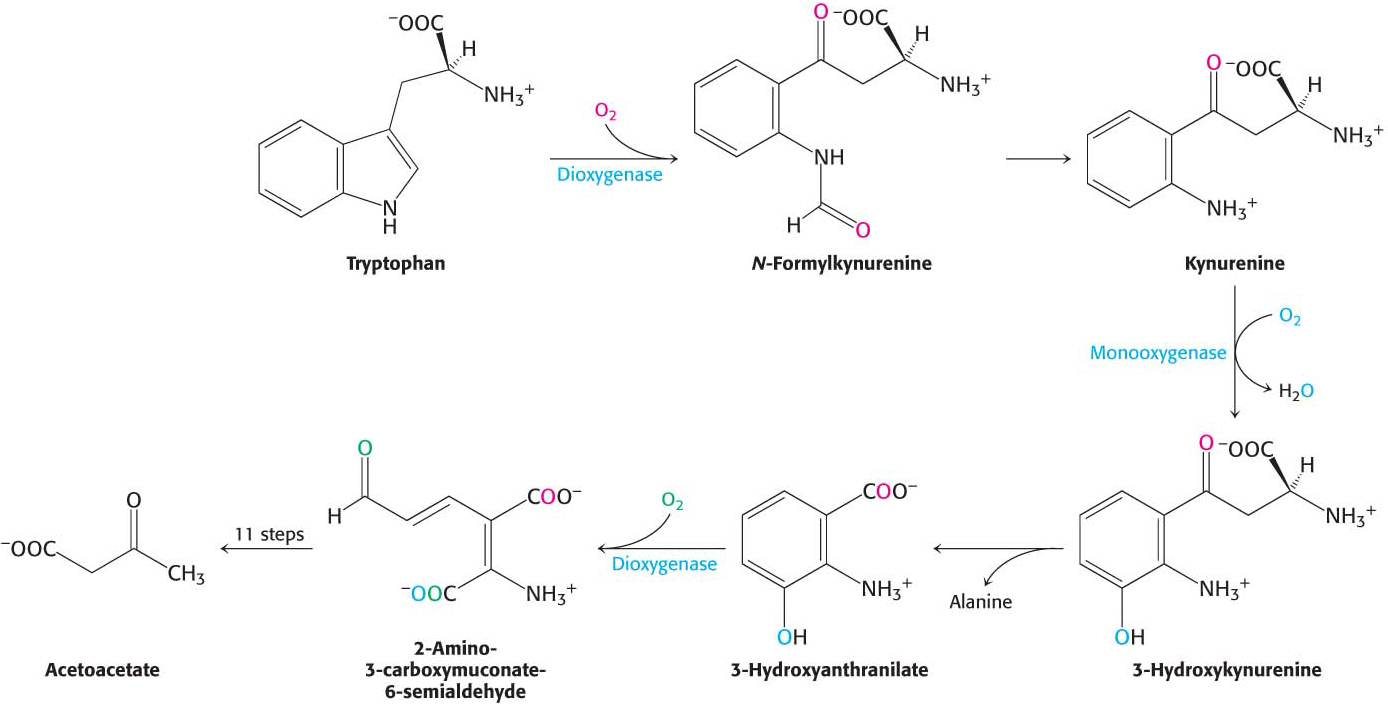
Methionine Is Degraded into Succinyl Coenzyme A
Methionine is converted into succinyl CoA in nine steps (Figure 30.13). The first step is the adenylation of methionine to form S-adenosylmethionine (SAM). Methyl donation and deadenylation yield homocysteine, which is metabolized to propionyl CoA, which, in turn, is processed to succinyl CoA, as was described above. Recall that S-adenosylmethionine plays a key role in cellular biochemistry as a common methyl donor in the cell (Chapter 29).
 CLINICAL INSIGHT
CLINICAL INSIGHTInborn Errors of Metabolism Can Disrupt Amino Acid Degradation
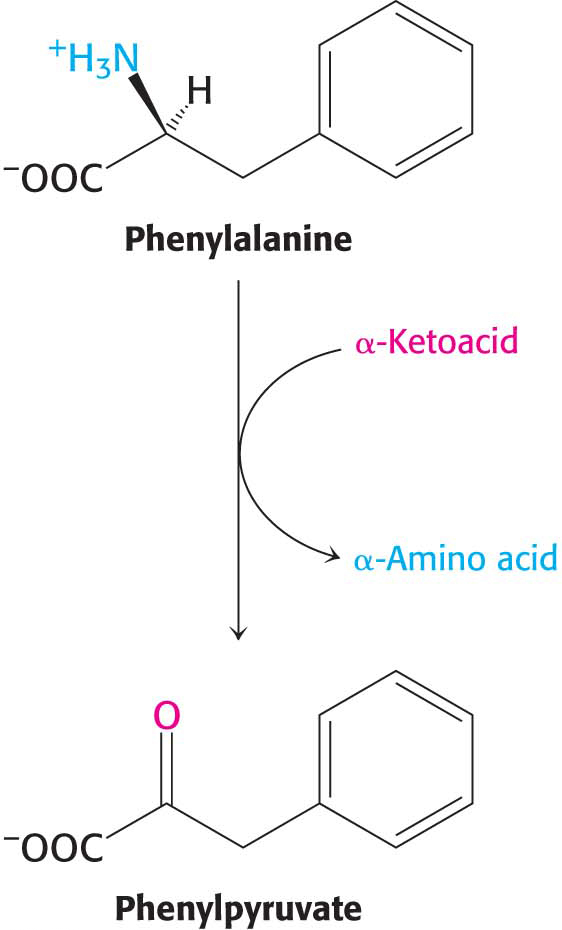
Errors in amino acid metabolism provided some of the first correlations between biochemical defects and pathological conditions (Table 30.1). Phenylketonuria, which occurs with a prevalence of 1 in 10,000 births, is perhaps the best known of the diseases of amino acid metabolism.

Phenylketonuria is caused by an absence or deficiency of phenylalanine hydroxylase or, more rarely, of its tetrahydrobiopterin cofactor. Phenylalanine accumulates in all body fluids because it cannot be converted into tyrosine for complete degradation. Normally, three-
Almost all untreated phenylketonurics are severely mentally retarded. The brain weight of these people is below normal, myelination of their nerves is defective, and their reflexes are hyperactive. The life expectancy of untreated phenylketonurics is drastically shortened. Half die by age 20 and, by age 30, three-
Phenylketonurics appear normal at birth but are severely defective by age 1 if untreated. The therapy for phenylketonuria is a low-

Early diagnosis of phenylketonuria is essential and has been accomplished by mass screening programs. The phenylalanine level in the blood is the preferred diagnostic criterion because it is more sensitive and reliable than the FeCl3 test. Prenatal diagnosis of phenylketonuria with DNA probes has become feasible because the gene has been cloned and the exact locations of many mutations have been discovered in the protein. Interestingly, whereas some mutations lower the activity of the enzyme, others decrease the enzyme concentration instead.
CLINICAL INSIGHTDetermining the Basis of the Neurological Symptoms of Phenylketonuria Is an Active Area of Research
The biochemical basis of retardation is not firmly established, but one hypothesis suggests that the lack of hydroxylase reduces the amount of tyrosine, an important precursor to neurotransmitters such as dopamine. Moreover, high concentrations of phenylalanine prevent amino acid transport of any tyrosine present as well as tryptophan, a precursor to the neurotransmitter serotonin, into the brain. Because all three of the amino acids are transported by the same carrier, phenylalanine will saturate the carrier, preventing access to tyrosine and tryptophan. Finally, high blood levels of phenylalanine result in higher levels of phenylalanine in the brain, and evidence suggests this elevated concentration inhibits glycolysis at pyruvate kinase, disrupts myelination of nerve fibers, and reduces the synthesis of several neurotransmitters.