PROBLEMS
Question 30.1
1. Keto counterparts. Name the α-ketoacid formed by the transamination of each of the following amino acids. ✓ 1
(a) Alanine
(b) Aspartate
(c) Glutamate
(d) Leucine
(e) Phenylalanine
(f) Tyrosine
Question 30.2
2. A versatile building block. (a) Write a balanced equation for the conversion of aspartate into glucose through the intermediate oxaloacetate. Which coenzymes participate in this transformation? (b) Write a balanced equation for the conversion of aspartate into oxaloacetate through the intermediate fumarate. ✓ 2
Question 30.3
3. Not very discriminating. Glutamate dehydrogenase is considered unusual in that it does not discriminate between NADH and NADPH, at least in some species. Explain why this failure to discriminate is unusual. ✓ 1
Question 30.4
4. Cooperation. How do aminotransferases and glutamate dehydrogenase cooperate in the metabolism of the amino group of amino acids? ✓ 1
Question 30.5
5. Taking away the nitrogen. What amino acids yield citric acid cycle components and glycolysis intermediates when deaminated? ✓ 1
Question 30.6
6. One reaction only. What amino acids can be deaminated directly? ✓ 1
Question 30.7
7. Nitrogen sources. What are the immediate biochemical sources for the two nitrogen atoms in urea? ✓ 1
Question 30.8
8. Counterparts. Match the biochemical on the right with the property on the left. ✓ 1
Formed from NH4+ Hydrolyzed to yield urea A second source of nitrogen Reacts with aspartate Cleavage yields fumarate Accepts the first nitrogen Final product | Carbamoyl phosphate Arginine Ornithine Aspartate Urea Argininosuccinate Citrulline |
Question 30.9
9. Completing the cycle. Four high-
Question 30.10
10. A good bet. A friend bets you a bazillion dollars that you can’t prove that the urea cycle is linked to the citric acid cycle and other metabolic pathways. Can you collect? ✓ 1
Question 30.11
11. A precise diagnosis. The result of a reaction between an infant’s urine and 2,4-
Question 30.12
12. Line up. Identify structures A–
Question 30.13
13. Sweet hazard. Why should phenylketonurics avoid the ingestion of aspartame, an artificial sweetener? (Hint: Aspartame is l-aspartyl-
Question 30.14
14. Déjà vu. N-Acetylglutamate is required as a cofactor in the synthesis of carbamoyl phosphate. How is N-acetylglutamate synthesized from glutamate? ✓ 1
Question 30.15
15. Precursors. Differentiate between ketogenic amino acids and glucogenic amino acids. ✓ 2
Question 30.16
16. Supply lines. The carbon skeletons of the 20 common amino acids can be degraded into a limited number of end products. What are the end products, and in what metabolic pathway are they commonly found? ✓ 2
Question 30.17
17. A sleight of hand. The end products of tryptophan are acetyl CoA and acetoacetyl CoA, yet tryptophan is a gluconeogenic amino acid in animals. Explain.
Question 30.18
18. Negative nitrogen balance. A deficiency of even one amino acid results in a negative nitrogen balance. In this state, more protein is degraded than is synthesized, and so more nitrogen is excreted than is ingested. Why would protein be degraded if one amino acid were missing?
Question 30.19
19. Argininosuccinic aciduria. Argininosuccinic aciduria is a condition that results when the urea-
Chapter Integration Problems
Question 30.20
20. Multiple substrates. In Chapter 8, we learned that there are two types of bisubstrate reactions, sequential and double-
Question 30.21
21. Closely related. Pyruvate dehydrogenase complex and α-ketoglutarate dehydrogenase complex are huge enzymes consisting of three discrete enzymatic activities. Which amino acids require a related enzyme complex for degradation, and what is the name of the enzyme? ✓ 2
Question 30.22
22. Ammonia toxicity. Glutamate is an important neurotransmitter whose levels must be carefully regulated in the brain. Explain how a high concentration of ammonia might disrupt this regulation. How might a high concentration of ammonia alter the citric acid cycle? ✓ 1
Question 30.23
23. Damaged liver. As stated in Chapter 28, excess alcoholic consumption can cause liver damage (cirrhosis). A consequence of liver damage is often ammonia poisoning. Explain. ✓ 1
Challenge Problems
Question 30.24
24. Inhibitor design. Compound A has been synthesized as a potential inhibitor of a urea-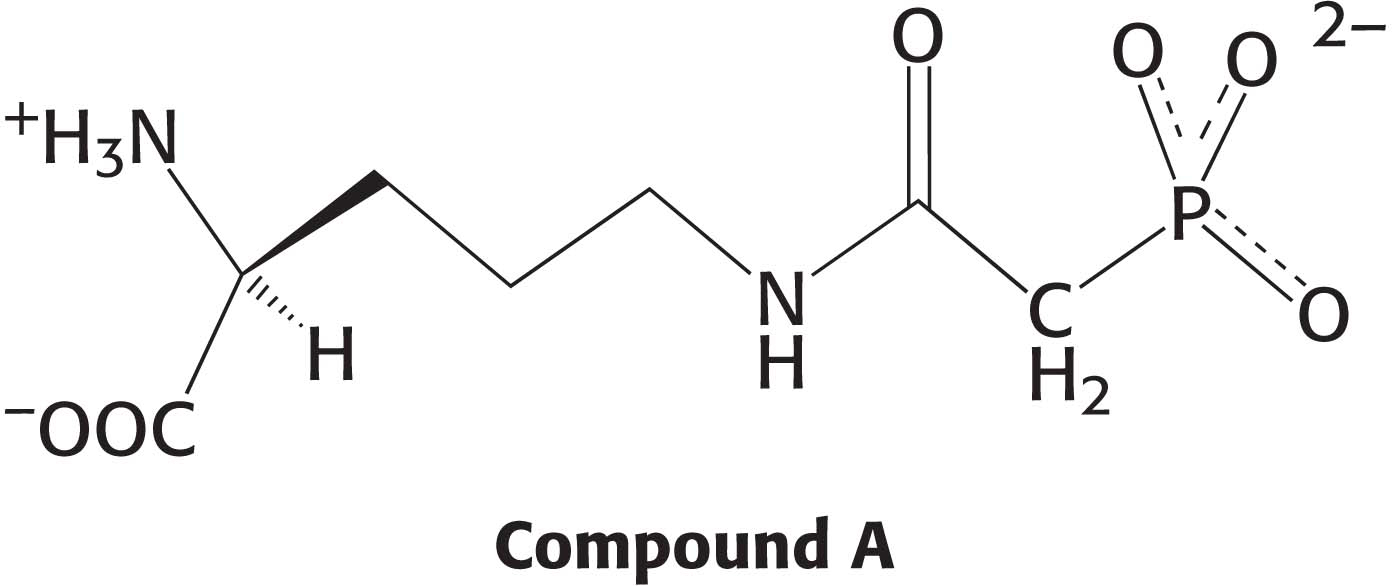
Which enzyme might compound A inhibit?
Question 30.25
25. Fuel choice. Within a few days after a fast begins, nitrogen excretion accelerates to a higher-
(a) What events trigger the initial surge of nitrogen excretion?
(b) Why does nitrogen excretion fall after several weeks of fasting?
(c) Explain the increase in nitrogen excretion when the lipid stores have been depleted.
Question 30.26
26. Isoleucine degradation. Isoleucine is degraded to acetyl CoA and succinyl CoA. Suggest a plausible reaction sequence, on the basis of reactions discussed in the text, for this degradation pathway. ✓ 2
Question 30.27
27. Enough cycles to have a race. The glucose–
Question 30.28
28. A serious situation. Pyruvate carboxylase deficiency is a fatal disorder. Patients with pyruvate carboxylase deficiency sometimes display some or all of the following symptoms: lactic acidosis, hyperammonemia (excess NH4+ in the blood), hypoglycemia, and demyelination of the regions of the brain due to insufficient lipid synthesis. Provide a possible biochemical rationale for each of these observations.
Selected Readings for this chapter can be found online at www.whfreeman.com/