39.1 The Genetic Code Links Nucleic Acid and Protein Information
✓ 1 Describe the genetic code.
For any sort of translation to take place, there must be a lexicon—
Three nucleotides encode an amino acid. Proteins are built from 20 amino acids, but there are only four bases in nucleic acids. Simple calculations show that a minimum of three bases is required to encode at least 20 amino acids. Genetic experiments showed that an amino acid is in fact encoded by a group of three bases, called a codon.
The code is nonoverlapping. Consider a base sequence ABCDEF. In an overlapping code, ABC specifies the first amino acid, BCD the next, CDE the next, and so on. In a nonoverlapping code, ABC designates the first amino acid, DEF the second, and so forth. Genetics experiments established the code to be nonoverlapping.
The code has no punctuation. In principle, one base (denoted as Q) might serve as a “comma” between codons:
… QABCQDEFQGHIQJKLQ …
However, it is not the case. Rather, the sequence of bases is read sequentially from a fixed starting point without punctuation.
The genetic code has directionality. The code is read from the 5′ end of the messenger RNA to its 3′ end.
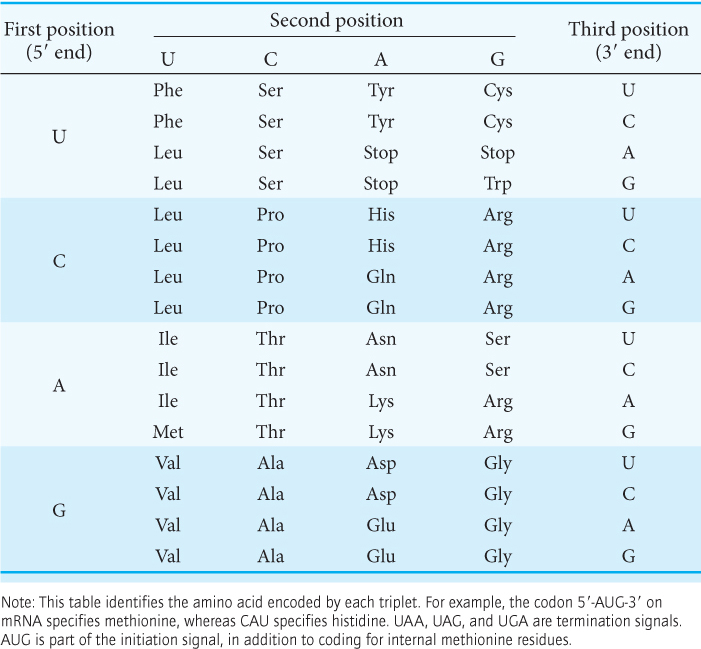
The genetic code is degenerate. In the context of the genetic code, degeneracy means that some amino acids are encoded by more than one codon, inasmuch as there are 64 possible base triplets and only 20 amino acids. In fact, 61 of the 64 possible triplets specify particular amino acids and 3 triplets (called Stop codons) designate the termination of translation. Thus, for most amino acids, there is more than one codon. Codons that specify the same amino acid are called synonyms. For example, CAU and CAC are synonyms for histidine.
All 64 codons have been deciphered (Table 39.1). Only tryptophan and methionine are encoded by just one triplet each. The other 18 amino acids are each encoded by two or more. Indeed, leucine, arginine, and serine are specified by six codons each.
What is the biological significance of the extensive degeneracy of the genetic code? If the code were not degenerate, 20 codons would designate amino acids and 44 would lead to chain termination. The probability of mutating to chain termination would therefore be much higher with a nondegenerate code. Chain-
The Genetic Code Is Nearly Universal
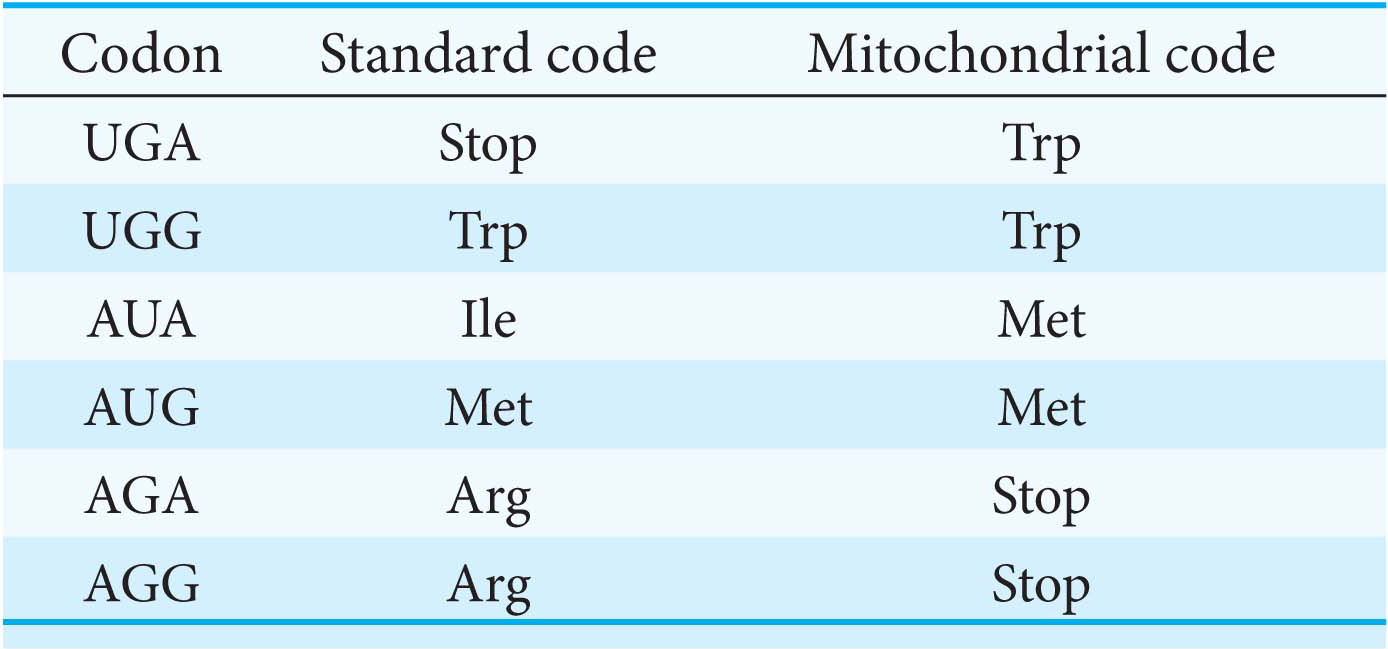
Most organisms use the same genetic code. This universality accounts for the fact that human proteins, such as insulin, can be synthesized in the bacterium E. coli and harvested from it for the treatment of diabetes. However, genome-
Transfer RNA Molecules Have a Common Design
The fidelity of protein synthesis requires the accurate recognition of three-
There is at least one tRNA molecule for each of the amino acids. These molecules have many common structural features, as might be expected because all tRNA molecules must be able to interact in nearly the same way with the ribosomes, mRNAs, and protein factors that participate in translation.
All known transfer RNA molecules have the following features:
Each is a single strand containing between 73 and 93 ribonucleotides (∼25 kDa).
The three-
dimensional molecule is L- shaped (Figure 39.1). 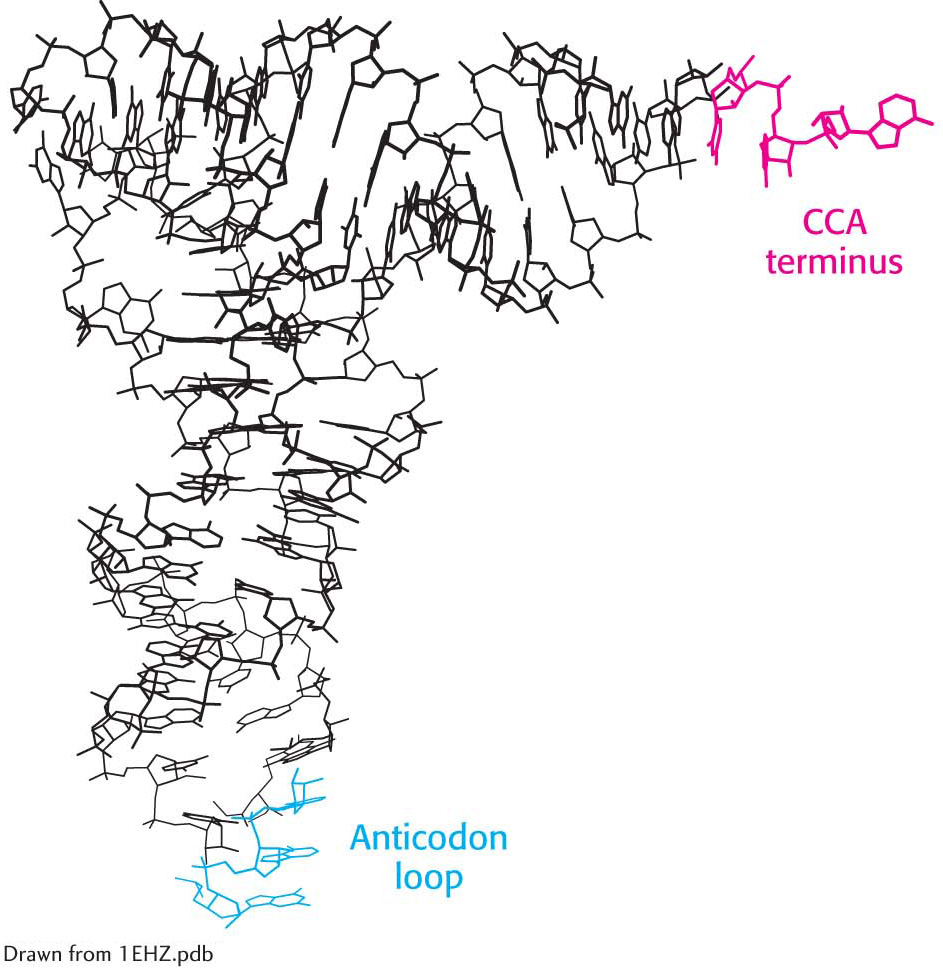 Figure 39.1
Figure 39.1 Transfer RNA structure. Notice the L-
Transfer RNA structure. Notice the L-shaped structure revealed by this skeletal model of yeast phenylalanyltRNA. The CCA region is at the end of one arm, and the anticodon loop is at the end of the other. Page 710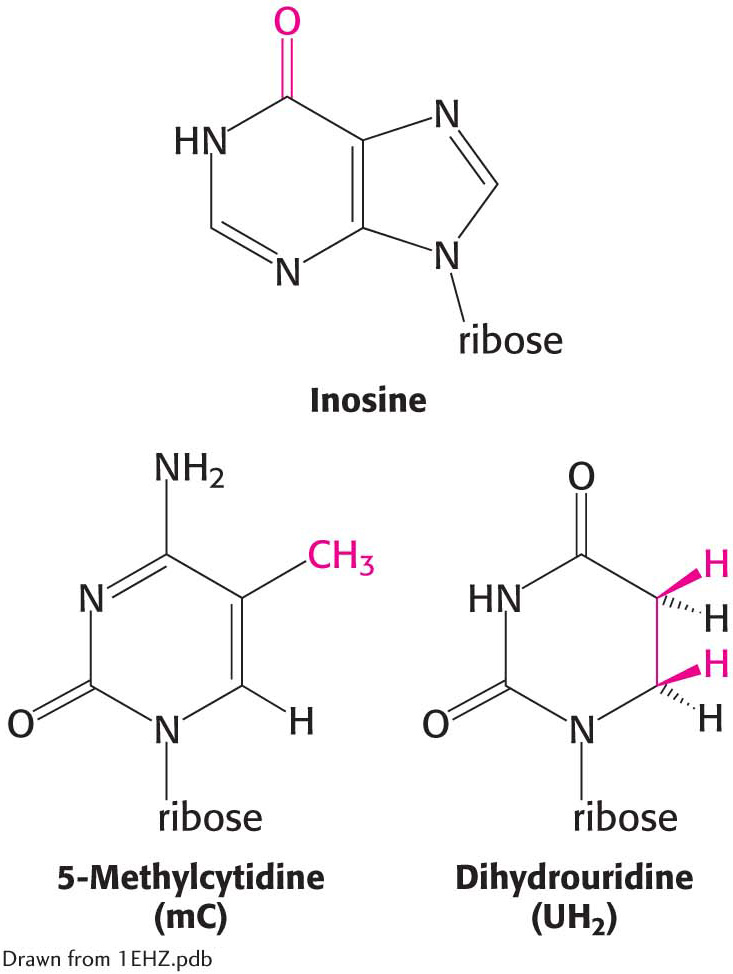
They contain many unusual bases, typically between 7 and 15 per tRNA. Some are methylated or dimethylated derivatives of A, U, C, and G. Methylation prevents the formation of certain base pairs, thereby rendering some of the bases accessible for interactions with other components of the translation machinery. In addition, methylation imparts a hydrophobic character to some regions of tRNAs, which may be important for their interaction with proteins required for protein synthesis. Modified bases, such as inosine, also are components of tRNA. The inosines in tRNA are formed by deamination of adenosine after the synthesis of the primary transcript.
When depicted on a two-
dimensional surface, all tRNA molecules can be arranged in a cloverleaf pattern, with about half the nucleotides in tRNAs base- paired to form double helices (Figure 39.2). Five groups of bases are not base- paired in this way: the 3′ CCA terminal region, which is part of a region called the acceptor stem; the TψC loop, which acquired its name from the sequence ribothymine- pseudouracil- cytosine; the “extra arm,” which contains a variable number of residues; the DHU loop, which contains several dihydrouracil residues; and the anticodon loop. The structural diversity generated by this combination of helices and loops containing modified bases ensures that the tRNAs can be uniquely distinguished, though structurally similar overall. 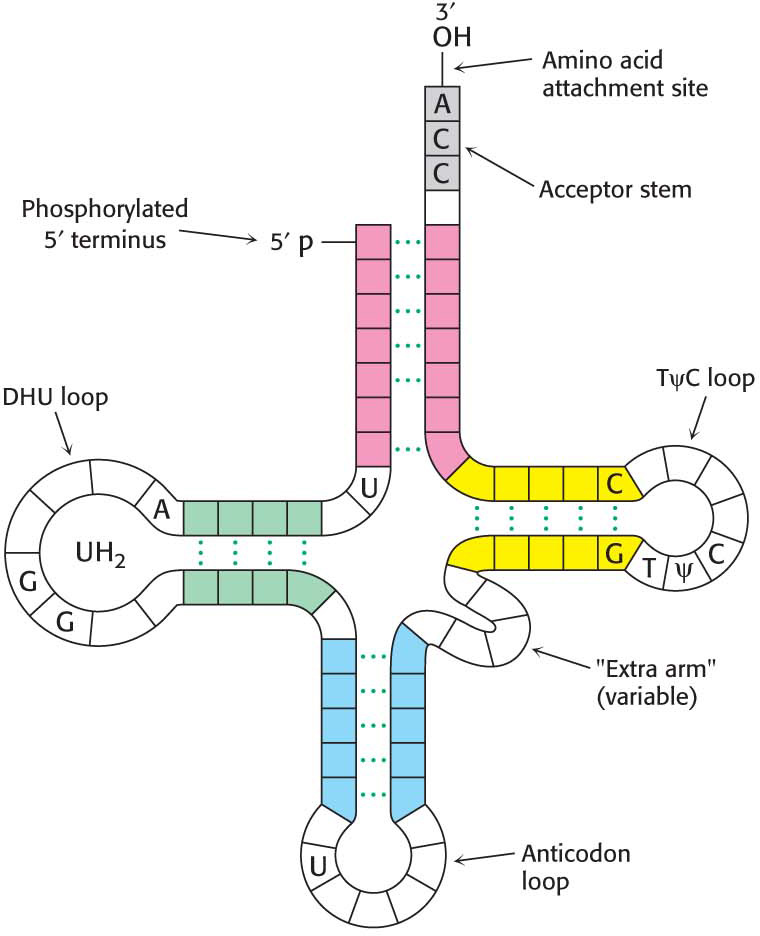 Figure 39.2 The general structure of transfer RNA molecules. The structure of the tRNA molecule is shown in the cloverleaf pattern. Comparison of the base sequences of many tRNAs reveals a number of conserved features.Page 711
Figure 39.2 The general structure of transfer RNA molecules. The structure of the tRNA molecule is shown in the cloverleaf pattern. Comparison of the base sequences of many tRNAs reveals a number of conserved features.Page 711The 5′ end of a tRNA is phosphorylated. The 5′ terminal residue is usually pG.
The activated amino acid is attached to a hydroxyl group of the adenosine residue located at the end of the 3′ CCA component of the acceptor stem. This region is a flexible single strand at the 3′ end of mature tRNAs.
The anticodon is present in a loop near the center of the sequence.
Some Transfer RNA Molecules Recognize More Than One Codon Because of Wobble in Base-Pairing
What are the rules that govern the recognition of a codon by the anticodon of a tRNA? A simple hypothesis is that each of the bases of the codon forms a Watson–
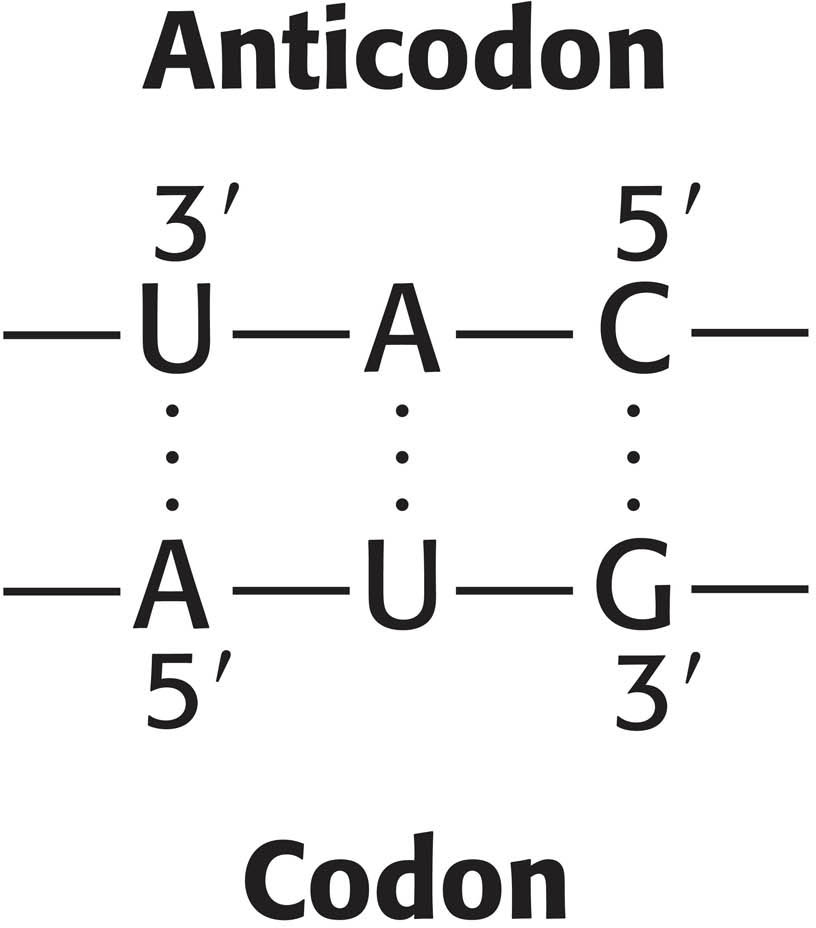
According to this model, a particular anticodon can recognize only one codon.
However, things are not so simple. Some tRNA molecules can recognize more than one codon. For example, consider the yeast alanyl-
Two generalizations concerning the codon–
The first two bases of a codon pair in the standard way. Recognition is precise. Hence, codons that differ in either of their first two bases must be recognized by different tRNAs. For example, both UUA and CUA encode leucine but are read by different tRNAs.
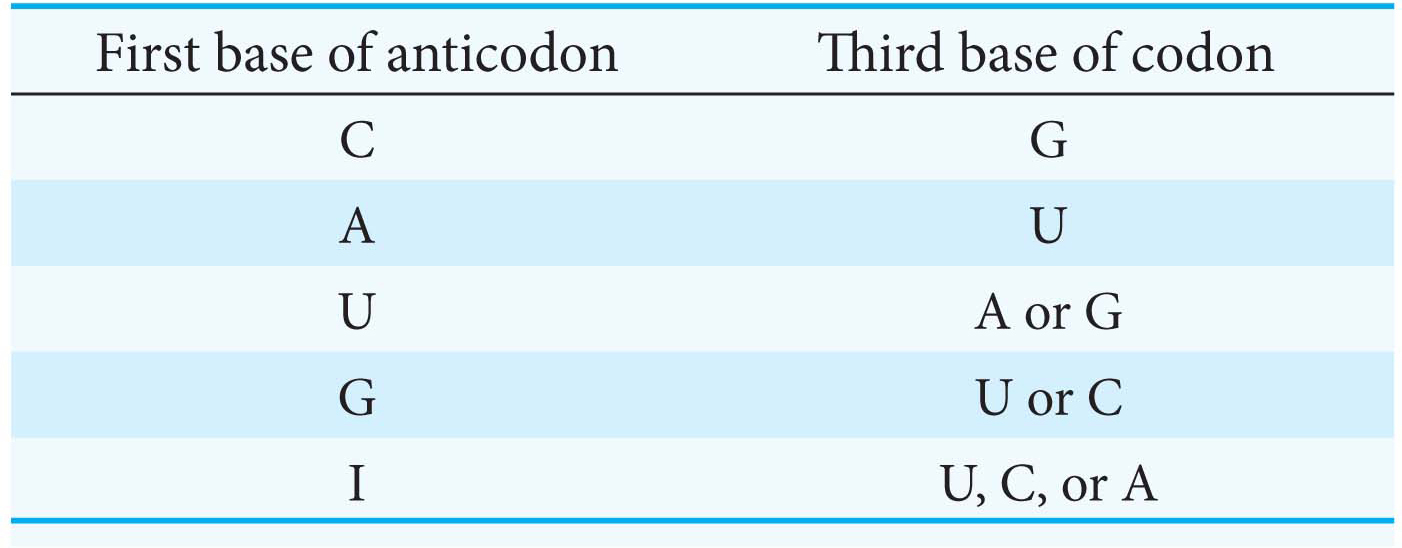
The first base of an anticodon determines whether a particular tRNA molecule reads one, two, or three kinds of codons: C or A (one codon), U or G (two codons), or I (three codons). Thus, part of the degeneracy of the genetic code arises from imprecision in the pairing of the third base of the codon with the first base of the anticodon. We see here a strong reason for the frequent appearance of inosine, one of the unusual nucleosides, in anticodons. Inosine maximizes the number of codons that can be read by a particular tRNA molecule.
The Synthesis of Long Proteins Requires a Low Error Frequency
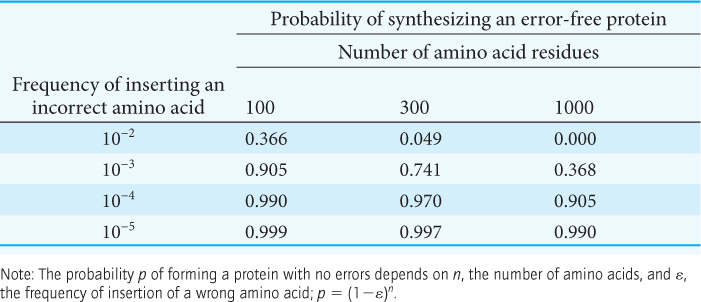
The process of transcription is analogous to copying, word for word, a page from a book. There is no change of alphabet or vocabulary; so the likelihood of a change in meaning is small. Translating the base sequence of an mRNA molecule into a sequence of amino acids is similar to translating the page of a book into another language. Translation is a complex process, entailing many steps and dozens of molecules. The potential for error exists at each step. The complexity of translation creates a conflict between two requirements: the process must be not only accurate, but also fast enough to meet a cell’s needs. How fast is “fast enough”? In E. coli, translation can take place at a rate of 40 amino acids per second, a truly impressive speed considering the complexity of the process.
How accurate must protein synthesis be? The average E. coli protein is about 300 amino acids in length, with several dozen greater than 1000 amino acids. Let us consider possible error rates when synthesizing proteins of this size. As Table 39.4 shows, an error frequency of 10−2 (one incorrect amino acid for every 100 correct ones incorporated into a protein) would be intolerable, even for small proteins. An error value of 10−3 would usually lead to the error-