4.5 The Amino Acid Sequence of a Protein Determines Its Three-Dimensional Structure
✓ 3 Describe the biochemical information that determines the final three-dimensional structure of proteins, and explain what powers the formation of this structure.
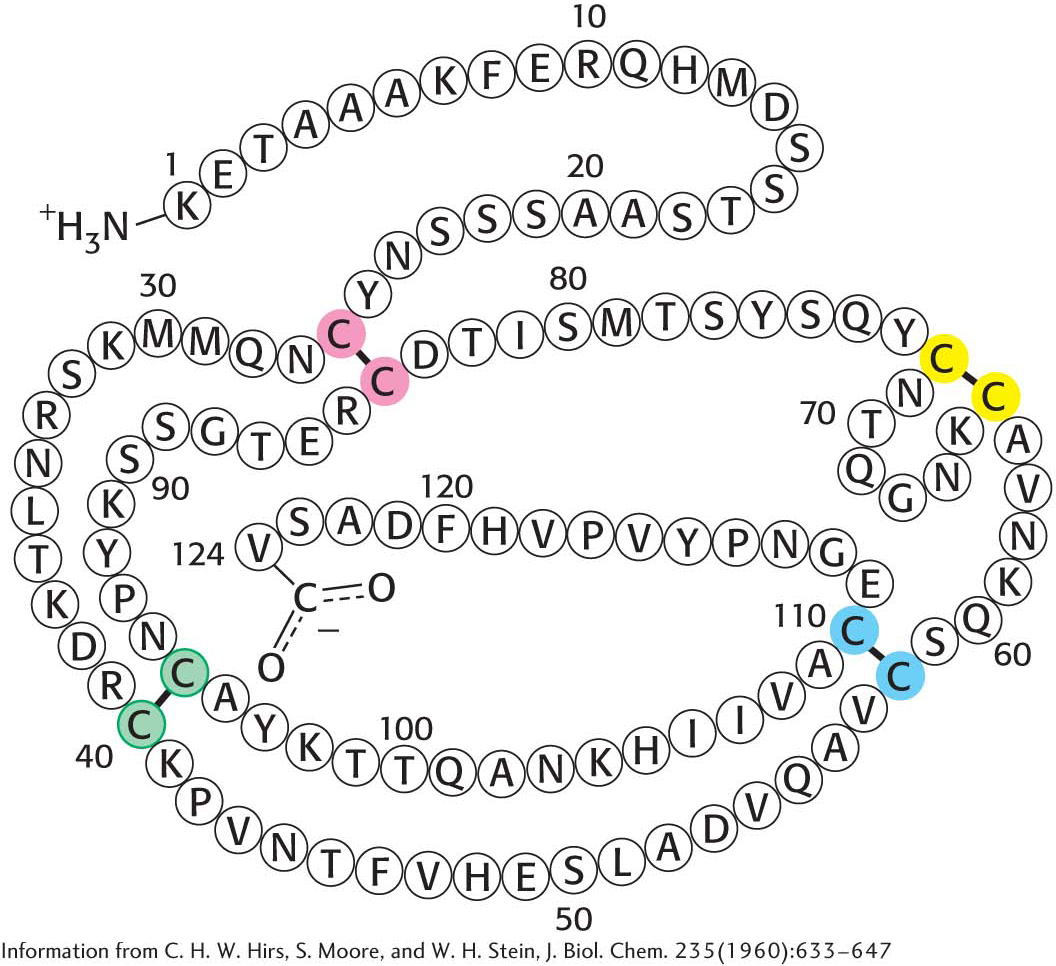
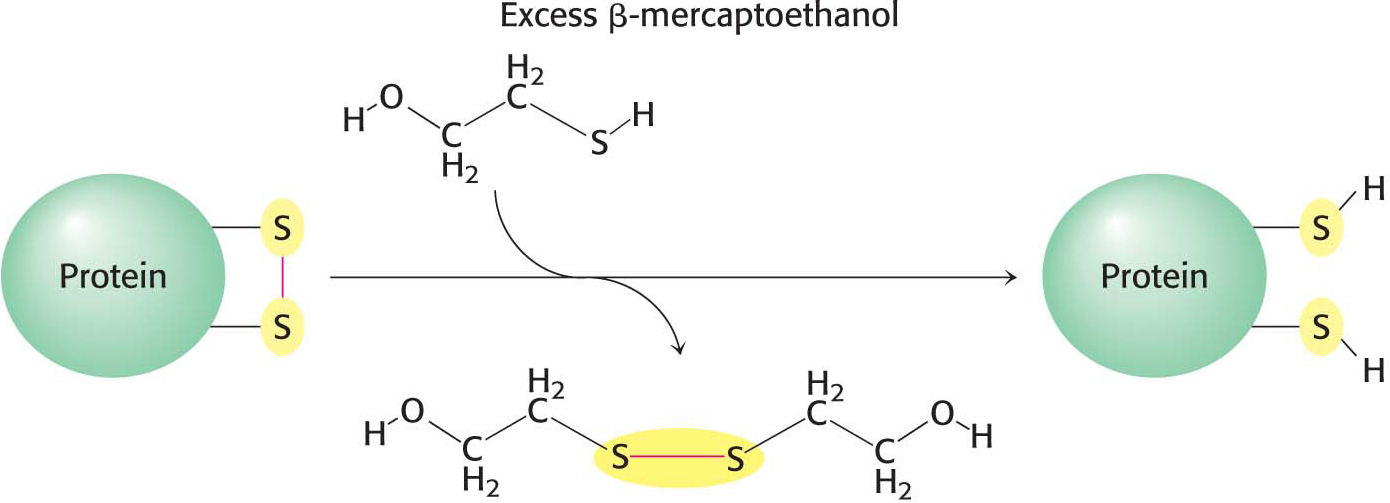
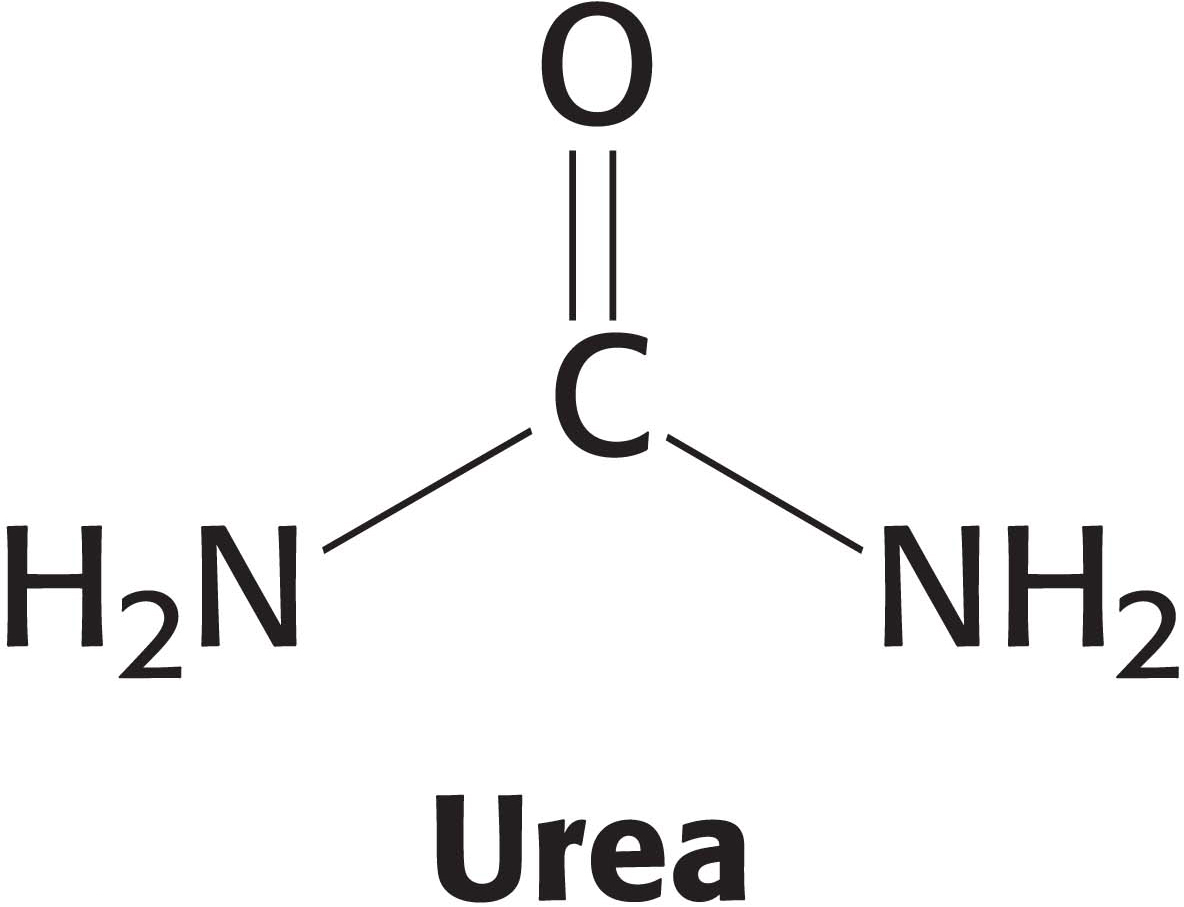
How is the elaborate three-dimensional structure of proteins attained? The classic work of Christian Anfinsen in the 1950s on the enzyme ribonuclease revealed the relation between the amino acid sequence of a protein and its conformation. Ribonuclease is a single polypeptide chain consisting of 124 amino acid residues cross-linked by four disulfide bonds (Figure 4.31). Anfinsen’s plan was to destroy the three-dimensional structure of the enzyme and to then determine the conditions required to restore the tertiary structure. Chaotropic agents, such as urea, which disrupt all of the noncovalent bonds in a protein, were added to a solution of the ribonuclease. The disulfide bonds where then cleaved reversibly with a sulfhydryl reagent such as β-mercaptoethanol (Figure 4.32). In the presence of a large excess of β-mercaptoethanol, the disulfides (cystines) are fully converted into sulfhydryls (cysteines).
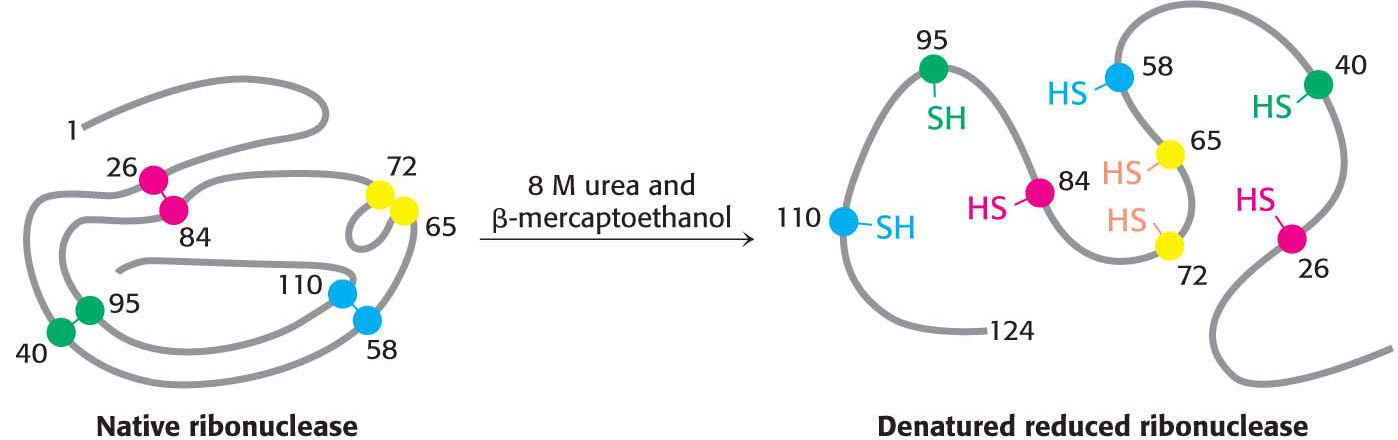
When ribonuclease was treated with β-mercaptoethanol in 8 M urea, the product was a randomly coiled polypeptide chain devoid of enzymatic activity. When a protein is converted into a randomly coiled peptide without its normal activity, it is said to be denatured (Figure 4.33).
Anfinsen then made the critical observation that the denatured ribonuclease, freed of urea and β-mercaptoethanol by dialysis, slowly regained enzymatic activity. He immediately perceived the significance of this finding: the enzyme spontaneously refolded into a catalytically active form with all of the correct disulfide bonds re-forming. All the measured physical and chemical properties of the refolded enzyme were virtually identical with those of the native enzyme. These experiments showed that the information needed to specify the catalytically active three-dimensional structure of ribonuclease is contained in its amino acid sequence. In other words, the native structure is the thermodynamically most stable structure. Subsequent studies have established the generality of this central principle of biochemistry: sequence specifies conformation. The dependence of conformation on sequence is especially significant because conformation determines function.
Page 61
Similar refolding experiments have been performed on many other proteins. In many cases, the native structure can be generated under suitable conditions. For other proteins, however, refolding does not proceed efficiently. In these cases, the unfolded protein molecules usually become tangled up with one another to form aggregates. Inside cells, proteins called chaperones block such illicit interactions.
Proteins Fold by the Progressive Stabilization of Intermediates Rather Than by Random Search
How does a protein make the transition from an unfolded structure to a unique conformation in the native form? One possibility is that all possible conformations are tried out to find the energetically most favorable one. How long would such a random search take? Cyrus Levinthal calculated that, if each residue of a 100-residue protein can assume three different conformations, the total number of structures would be 3100, which is equal to 5 × 1047. If the conversion of one structure into another were to take 10−13 seconds (s), the total search time would be 5 × 1047 × 10−13 s, which is equal to 5 × 1034 s, or 1.6 × 1027 years. Clearly, it would take much too long for even a small protein to fold properly by randomly trying out all possible conformations. Moreover, Anfinsen’s experiments showed that proteins do fold on a much more limited time scale. The enormous difference between calculated and actual folding times is called Levinthal’s paradox. Levinthal’s paradox and Anfinsen’s results suggest that proteins do not fold by trying every possible conformation; rather, they must follow at least a partly defined folding pathway consisting of intermediates between the fully denatured protein and its native structure.
The way out of this paradox is to recognize the power of cumulative selection. Richard Dawkins, in his book The Blind Watchmaker, asked how long it would take a monkey poking randomly at a typewriter to reproduce Hamlet’s remark to Polonius, “Methinks it is like a weasel” (Figure 4.34). An astronomically large number of keystrokes, of the order of 1040, would be required. However, suppose that we preserved each correct character and allowed the monkey to retype only the wrong ones. In this case, only a few thousand keystrokes, on average, would be needed. The crucial difference between these cases is that the first employs a completely random search, whereas, in the second, partly correct intermediates are retained.
The essence of protein folding is the tendency to retain partly correct intermediates because they are slightly more stable than unfolded regions. However, the protein-folding problem is much more difficult than the one presented to our simian Shakespeare. First, the criterion of correctness is not a residue-by-residue scrutiny of conformation by an omniscient observer but rather the total free energy of the folding intermediate. Second, even correctly folded proteins are only marginally stable. The free-energy difference between the folded and the unfolded states of a typical 100-residue protein is 42 kJ mol−1 (10 kcal mol−1); thus, each residue contributes on average only 0.42 kJ mol−1 (0.1 kcal mol−1) of energy to maintain the folded state. This amount is less than the amount of thermal energy, which is 2.5 kJ mol−1 (0.6 kcal mol−1) at room temperature. This meager stabilization energy means that correct intermediates, especially those formed early in folding, can be lost. Nonetheless, the interactions that lead to folding can stabilize intermediates as structure builds up. The analogy is that the monkey would be somewhat free to undo its correct keystrokes.
Page 62
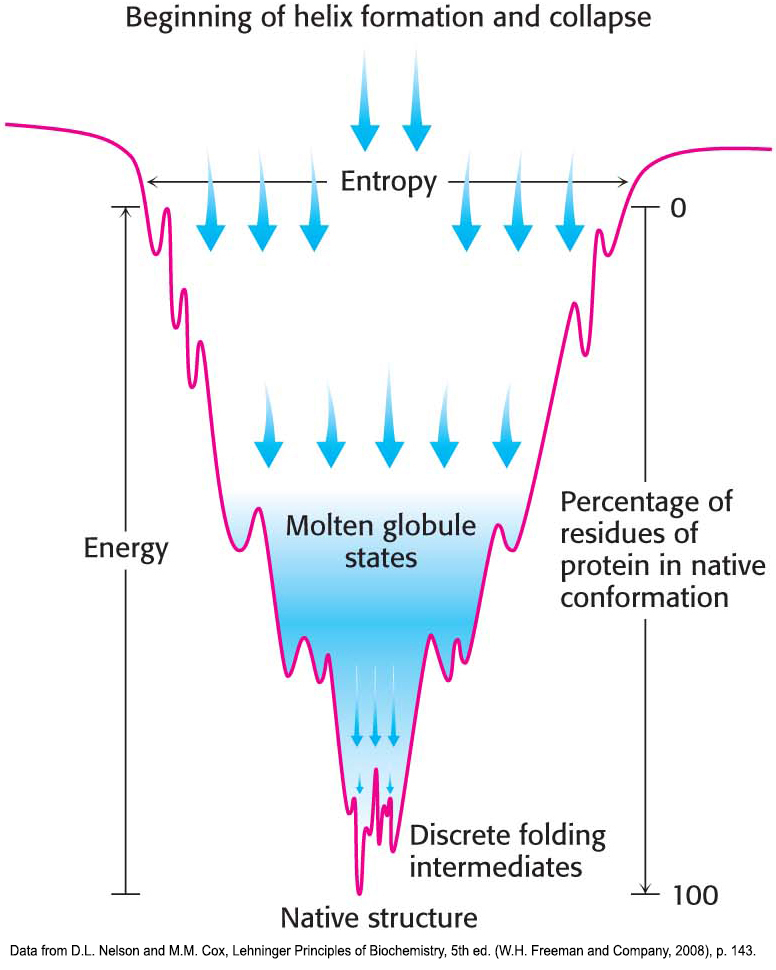
The folding of proteins is sometimes visualized as a folding funnel, or energy landscape (Figure 4.35). The breadth of the funnel represents all possible conformations of the unfolded protein. The depth of the funnel represents the energy difference between the unfolded and the native protein. Each point on the surface represents a possible three-dimensional structure and its energy value. The funnel suggests that there are alternative pathways to the native, or most stable, structure.
One model pathway postulates that local interactions take place first—in other words, secondary structure forms—and these secondary structures facilitate the long-range interactions leading to tertiary-structure formation. Another model pathway proposes that the hydrophobic effect brings together hydrophobic amino acids that are far apart in the amino acid sequence. The drawing together of hydrophobic amino acids in the interior leads to the formation of a globular structure. Because the hydrophobic interactions are presumed to be dynamic, allowing the protein to form progressively more stable interactions, the structure is called a molten globule. Another, more general model, called the nucleation–condensation model, is essentially a combination of the two preceding models. In the nucleation–condensation model, both local and long-range interactions take place to lead to the formation of the native state.
Some Proteins Are Inherently Unstructured and Can Exist in Multiple Conformations
The discussion of protein folding thus far is based on the paradigm that a given protein amino acid sequence will fold into a particular three-dimensional structure. This paradigm holds well for many proteins, such as enzymes and transport proteins. However, it has been known for some time that some proteins can adopt two different structures, one of which results in protein aggregation and pathological conditions. Such alternate structures originating from a unique amino acid sequence were thought to be rare: the exception to the paradigm. However, recent work has called into question the universality of the idea that each amino acid sequence gives rise to one structure for certain proteins, even under normal cellular conditions.
Our first example is a class of proteins referred to as intrinsically unstructured proteins (IUPs). As the name suggests, these proteins, completely or in part, do not have a discrete three-dimensional structure under physiological conditions. Indeed, an estimated 50% of eukaryotic proteins have at least one unstructured region greater than 30 amino acids in length. These proteins assume a defined structure on interaction with other proteins. This molecular versatility means that one protein can assume different structures and interact with different partners, yielding different biochemical functions. IUPs appear to be especially important in signaling and regulatory pathways.
Page 63
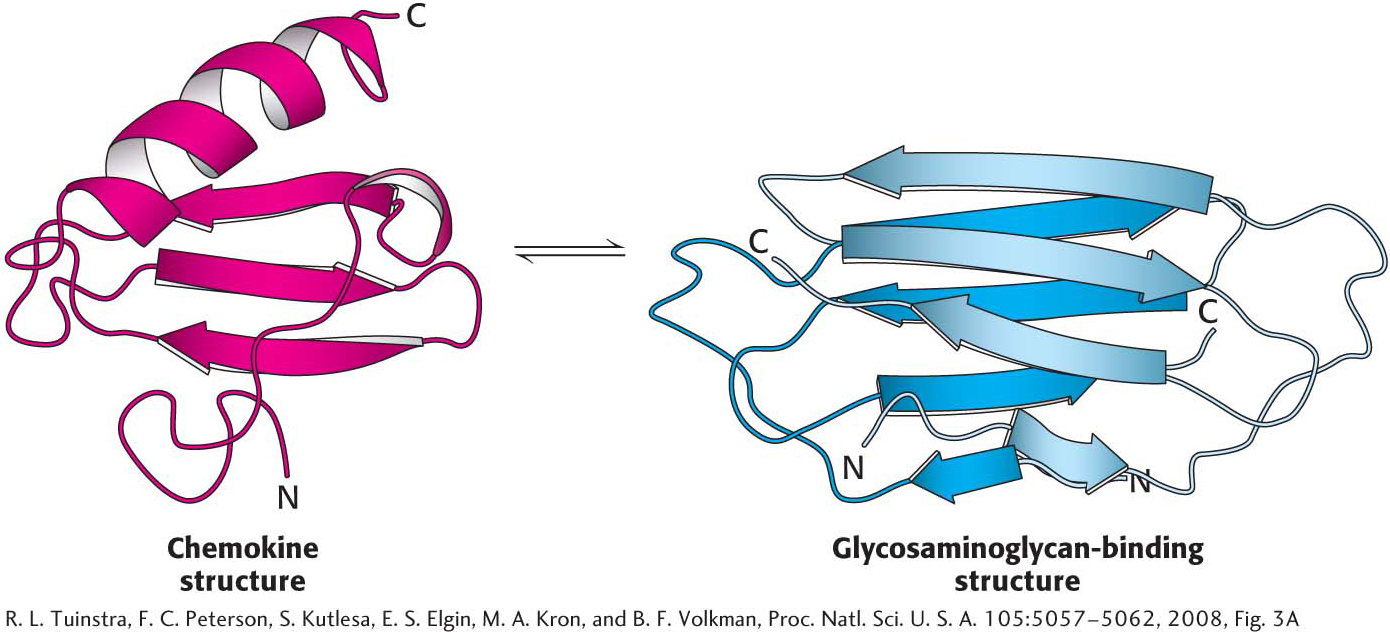
Another class of proteins that do not adhere to the paradigm are metamorphic proteins. These proteins appear to exist in an ensemble of structures of approximately equal energy that are in equilibrium. Small molecules or other proteins may bind to a particular member of the ensemble, resulting in a complex having a biochemical function that differs from that of another complex formed by the same metamorphic protein bound to a different partner. An especially clear example of a metamorphic protein is the cytokine lymphotactin. Cytokines are signal molecules in the immune system that bind to receptor proteins on the surface of immune-system cells, instigating an immunological response. Lymphotactin exists in two very different structures that are in equilibrium (Figure 4.36). One structure is a characteristic of chemokines, consisting of a three-stranded β sheet and a carboxyl-terminal helix. This structure binds to its receptor and activates it. The alternative structure is an identical dimer of all β sheets. When in this structure, lymphotactin binds to glycosaminglycan, a complex carbohydrate (Chapter 10). The biochemical activities of each structure are mutually exclusive: the cytokine structure cannot bind the glycosaminoglycan, and the β-sheet structure cannot activate the receptor. Yet, remarkably, both activities are required for full biochemical activity of the cytokine.
Note that IUPs and metamorphic proteins effectively expand the protein-encoding capacity of the genome. In some cases, a gene can encode a single protein that has more than one structure and function. These examples also illustrate the dynamic nature of the study of biochemistry and its inherent excitement: even well-established ideas are often subject to modifications.
 CLINICAL INSIGHT
CLINICAL INSIGHTProtein Misfolding and Aggregation Are Associated with Some Neurological Diseases
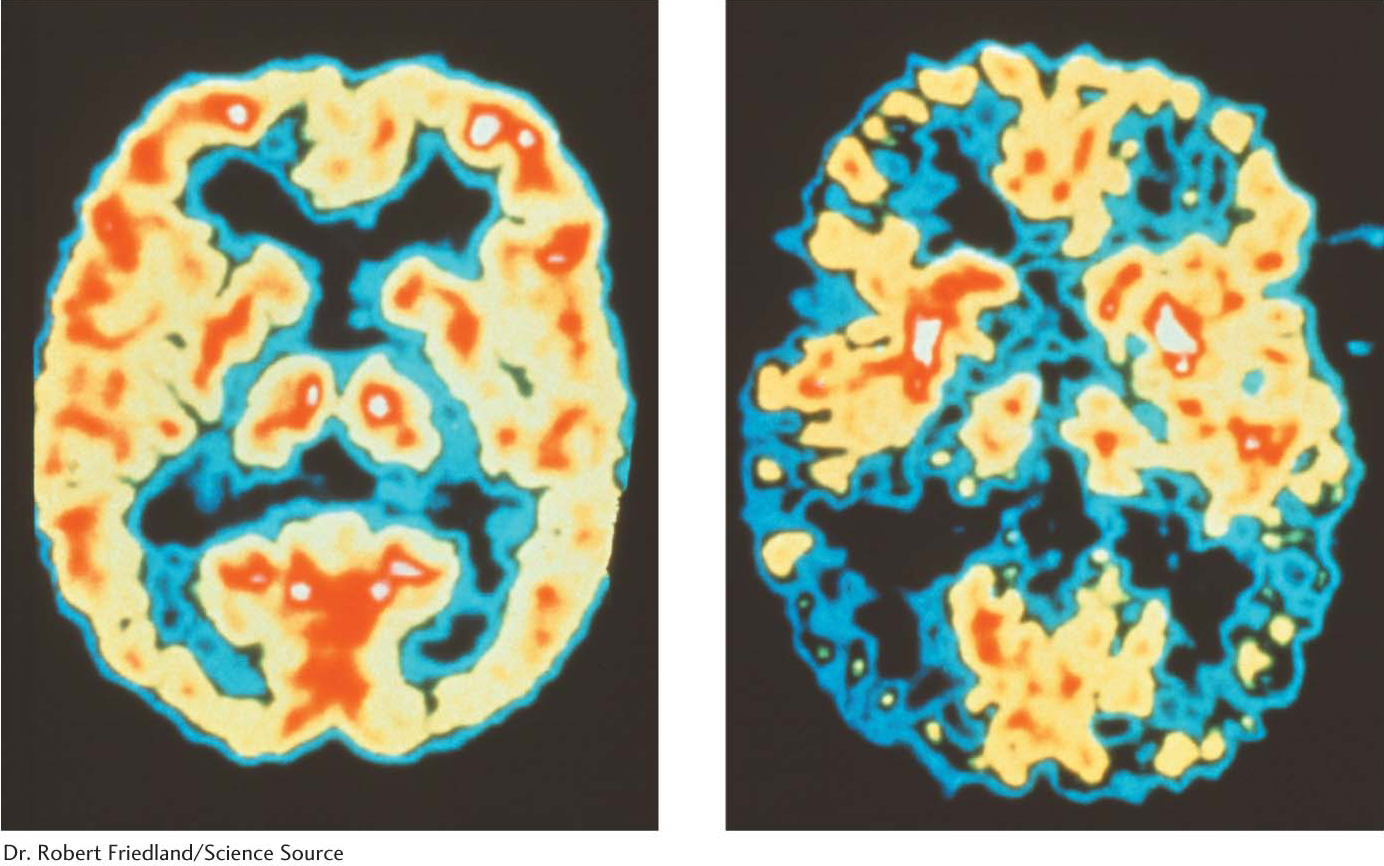
Understanding protein folding and misfolding is of more than academic interest. A host of diseases, including Alzheimer disease, Parkinson disease, Huntington disease, and transmissible spongiform encephalopathies (prion disease), are associated with improperly folded proteins. All of these diseases result in the deposition of protein aggregates, called amyloid fibrils or plaques (Figure 4.37). These diseases are consequently referred to as amyloidoses. A common feature of amyloidoses is that normally soluble proteins are converted into insoluble fibrils rich in β sheets. The correctly folded protein is only marginally more stable than the incorrect form. But the incorrect forms aggregates, pulling more correct forms into the incorrect form. We will focus on the transmissible spongiform encephalopathies.
One of the great surprises in modern medicine was the discovery by Stanley Prusiner that certain infectious neurological diseases were found to be transmitted by agents that were similar in size to viruses but consisted only of protein. These diseases include bovine spongiform encephalopathy (commonly referred to as mad cow disease) and the analogous diseases in other organisms, including Creutzfeldt–Jakob disease (CJD) in human beings and scrapie in sheep. The agents causing these diseases are termed prions. Prions are composed largely or completely of a cellular protein called PrP, which is normally present in the brain. The prions are aggregated forms of the PrP protein termed PrPSC.
Page 64
The structure of the normal protein PrP contains extensive regions of α helix and relatively little β-strand structure. The structure of a mammalian PrPSC has not yet been determined, because of challenges posed by its insoluble and heterogeneous nature. However, various evidence indicates that some parts of the protein that had been in α-helical or turn conformations have been converted into β-strand conformations. This conversion suggests that the PrP is only slightly more stable than the β-strand-rich PrPSC; however, after the PrPSC has formed, the β strands of one protein link with those of another to form β sheets, joining the two proteins and leading to the formation of aggregates, or amyloid fibrils.
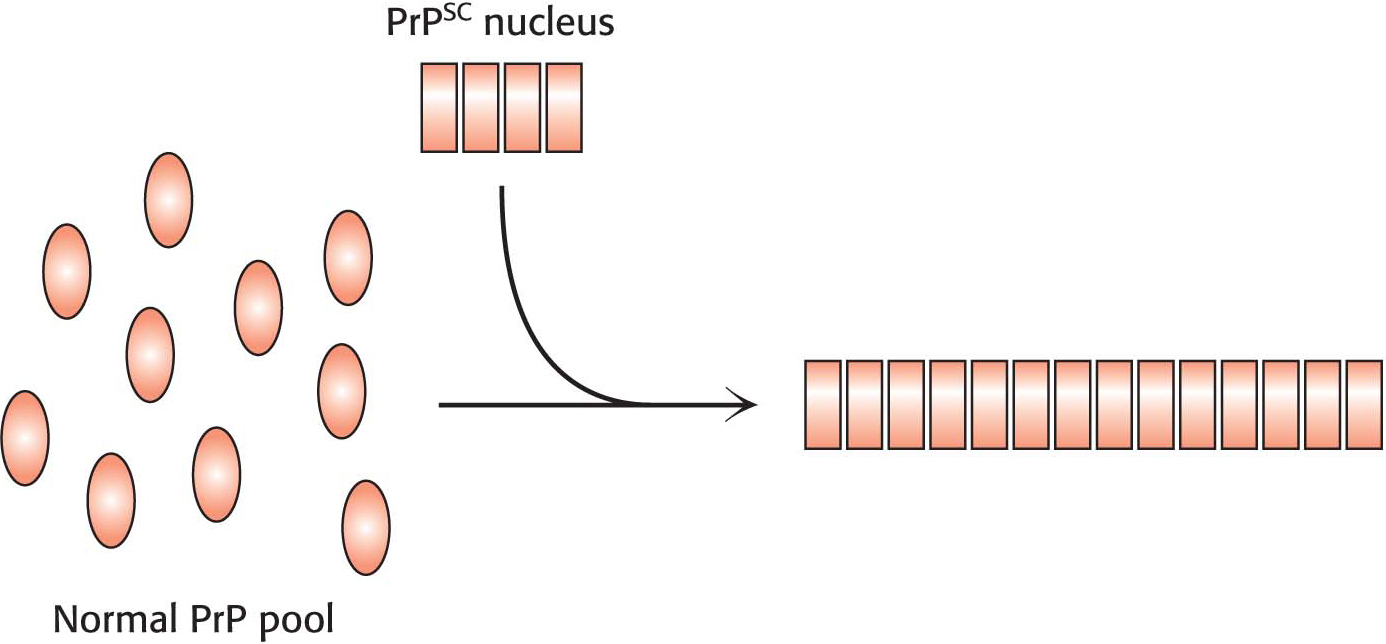
With the realization that the infectious agent in prion diseases is an aggregated form of a protein that is already present in the brain, a model for disease transmission emerges (Figure 4.38). Protein aggregates built of abnormal forms of PrP act as nuclei to which other PrP molecules attach. Prion diseases can thus be transferred from one individual organism to another through the transfer of an aggregated nucleus, as likely happened in the mad cow disease outbreak in the United Kingdom in the 1990s: cattle given animal feed containing material from diseased cows developed the disease in turn. Amyloid fibers are also seen in the brains of patients with certain noninfectious neurodegenerative diseases such as Alzheimer and Parkinson diseases. How such aggregates lead to the death of the cells that harbor them is an active area of research.
Page 65