
5.4 Determination of Primary Structure Facilitates an Understanding of Protein Function
Now that we have purified our protein, be it lactate dehydrogenase or the estrogen receptor, what is the next step in learning about the protein? An important means of characterizing a pure protein is to determine its primary structure, which can tell us much about the protein. Recall that the primary structure of a protein is the determinant of its three-
Let us examine first how we can sequence a simple peptide, such as
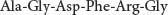
The first step is to determine the amino acid composition of the peptide. The peptide is hydrolyzed into its constituent amino acids by heating it in strong acid. The individual amino acids can then be separated by ion-
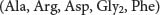
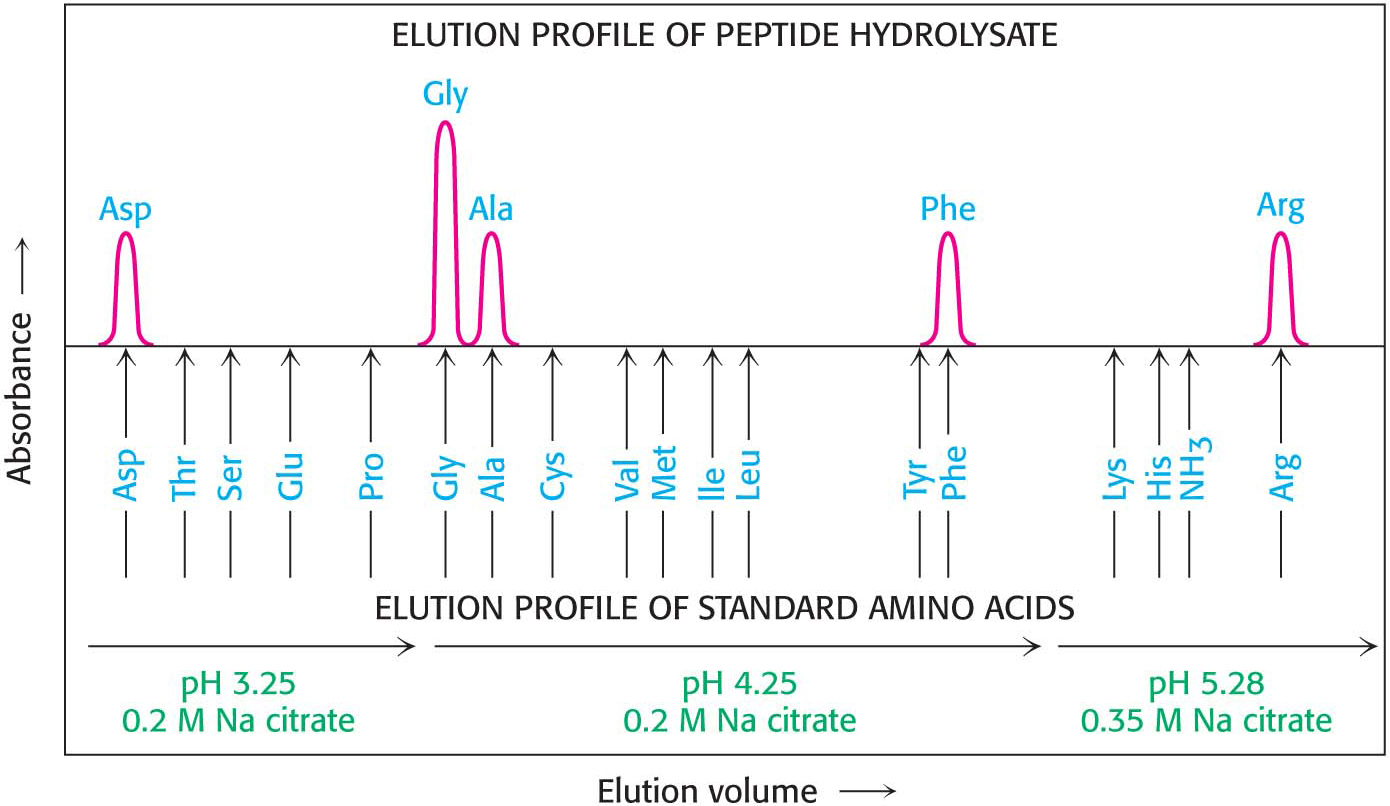
The parentheses denote that this is the amino acid composition of the peptide, not its sequence.
The sequence of a protein can then be determined by a process called the Edman degradation. The Edman degradation sequentially removes one residue at a time from the amino end of a peptide (Figure 5.26). Phenyl isothiocyanate reacts with the terminal amino group of the peptide, which then cyclizes and breaks off the peptide, yielding an intact peptide shortened by one amino acid. The cyclic compound is a phenylthiohydantoin (PTH)–amino acid, which can be identified by chromatographic procedures. The Edman procedure can then be repeated sequentially to yield the amino acid sequence of the peptide.
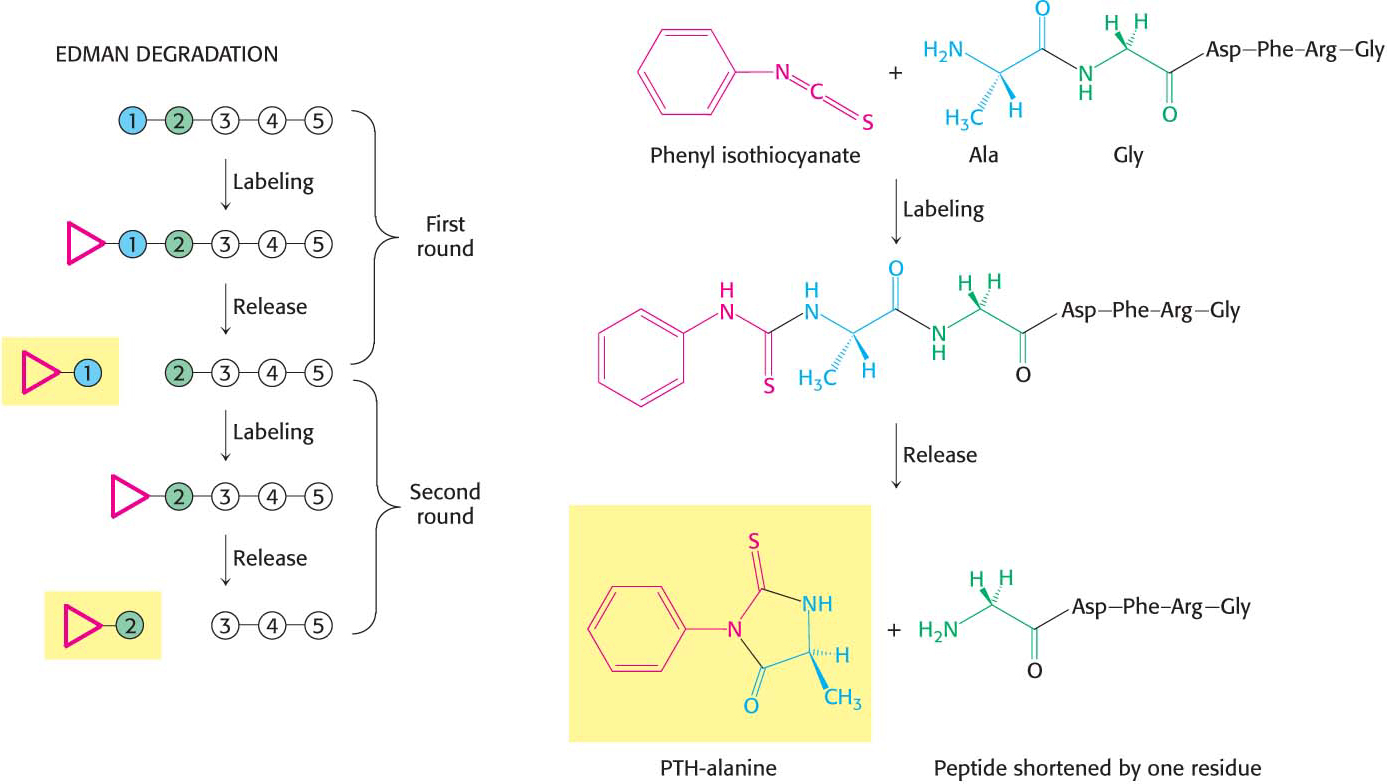
In principle, we should be able to sequence an entire protein by using the Edman method. In practice, the peptides cannot be much longer than about 50 residues, because the reactions of the Edman method are not 100% efficient and, eventually, the sequencing reactions are out of order. We can circumvent this obstacle by cleaving the original protein at specific amino acids into smaller peptides that can be sequenced independently. In essence, the strategy is to divide and conquer.
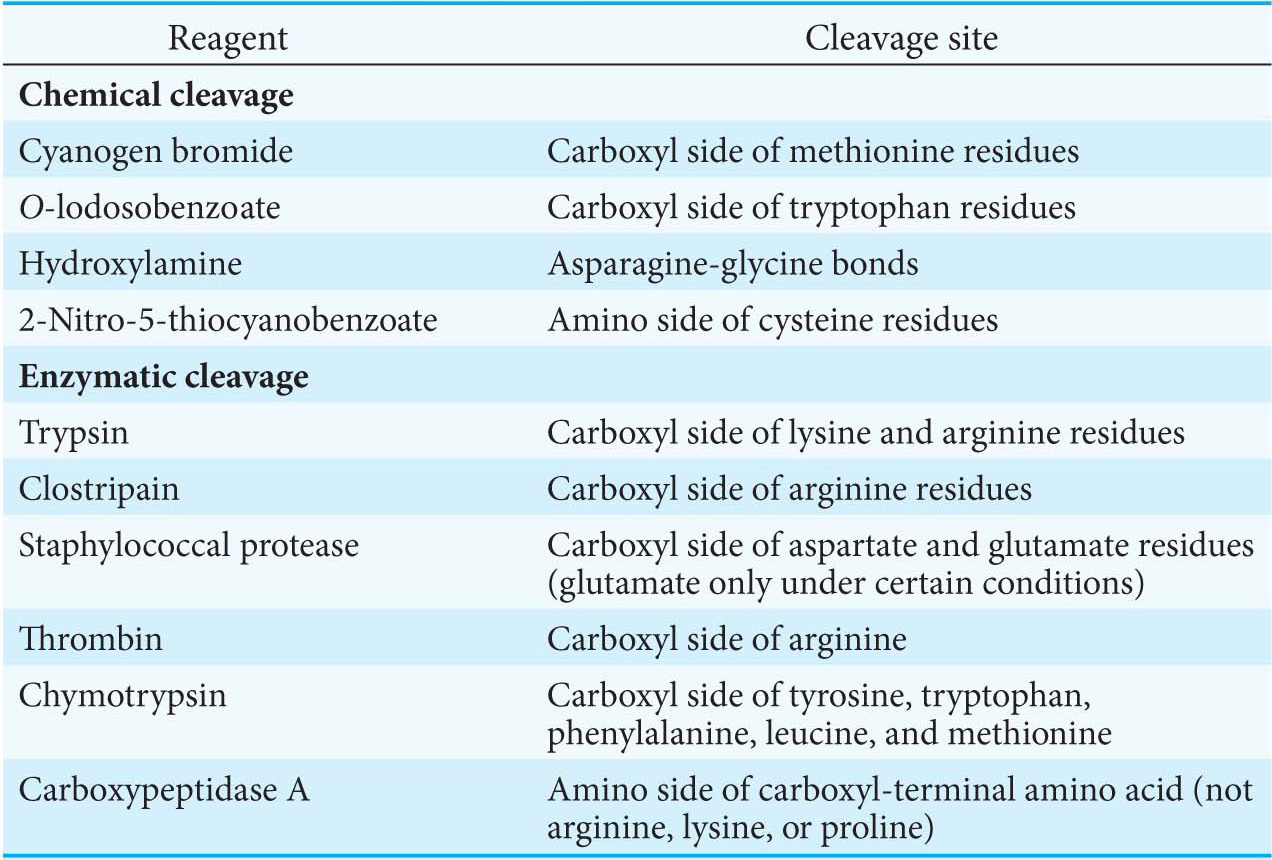
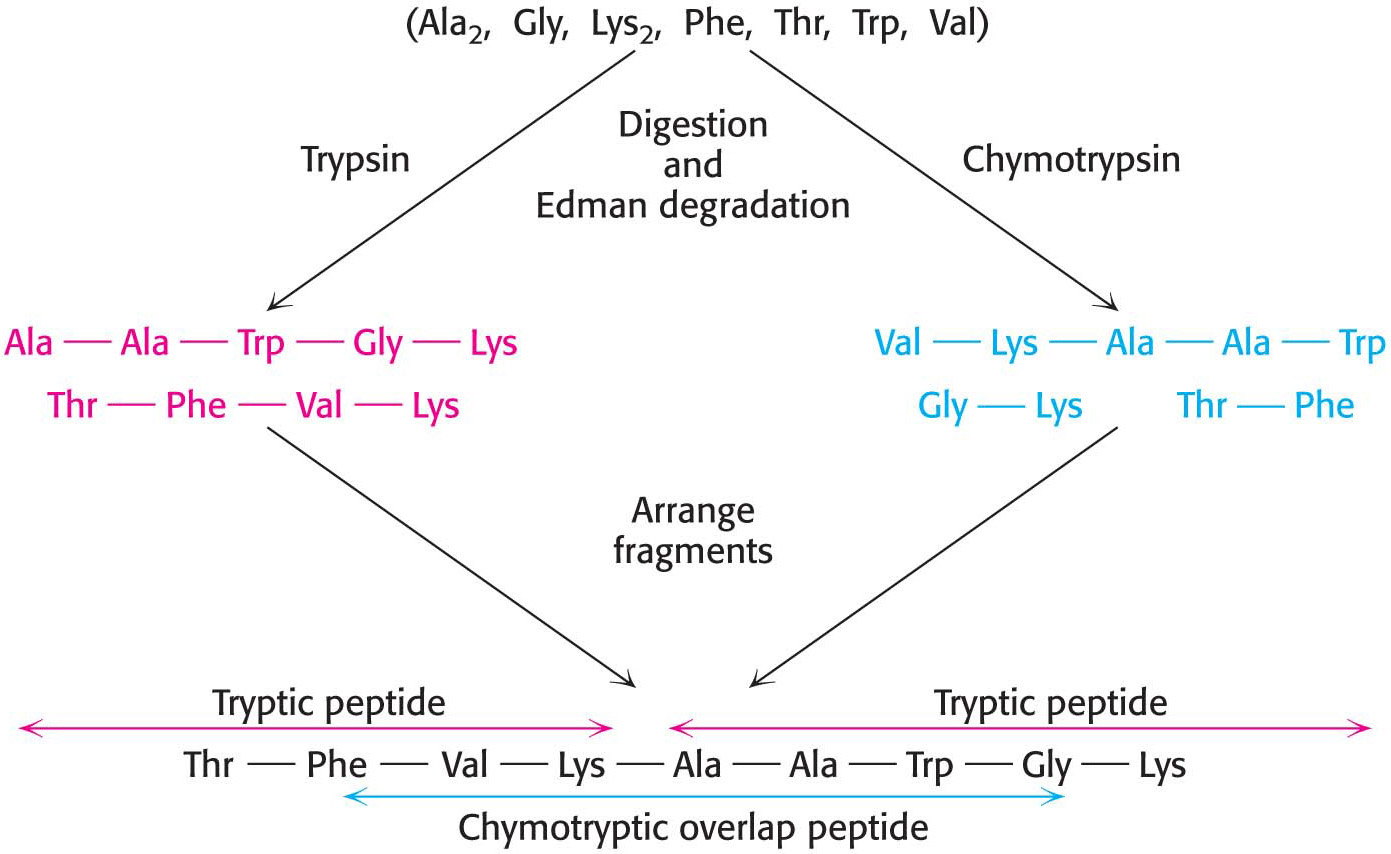
Specific cleavage can be achieved by chemical or enzymatic methods.Table 5.3 gives several ways of specifically cleaving polypeptide chains. The peptides obtained by specific chemical or enzymatic cleavage are separated, and the sequence of each purified peptide is then determined by the Edman method. At this point, the amino acid sequences of segments of the protein are known, but the order of these segments is not yet defined. How can we order the peptides to obtain the primary structure of the original protein? The necessary additional information is obtained from overlap peptides (Figure 5.27). A second cleavage technique is used to split the polypeptide chain at different sites. Some of the peptides from the second cleavage will overlap two or more peptides from the first cleavage, and they can be used to establish the order of the peptides. The entire amino acid sequence of the polypeptide chain is then known.
Mass Spectrometry Can Be Used to Determine a Protein’s Mass, Identity, and Sequence
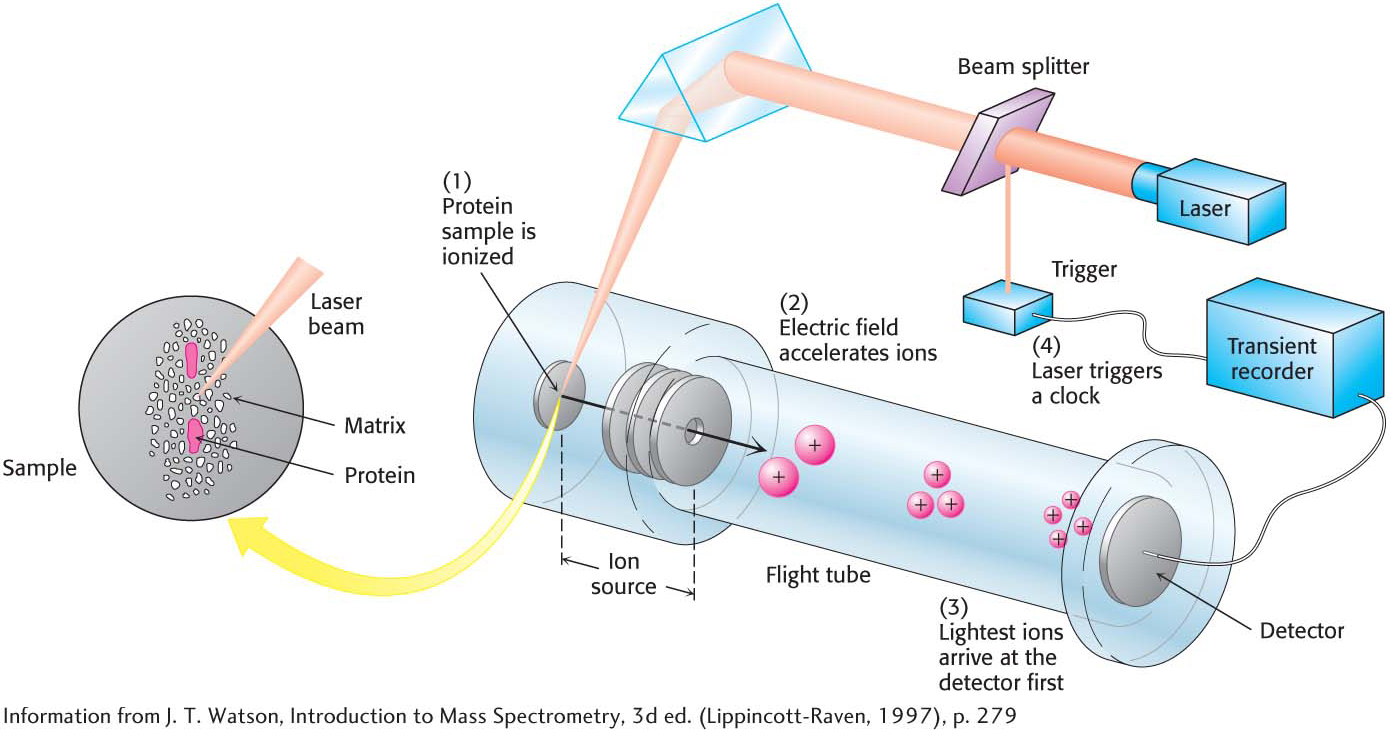
Although Edman degradation has provided a wealth of sequence information, it has largely been supplanted by the powerful technique of mass spectrometry. Before we can examine how mass spectrometry can be used to sequence a protein, we will investigate how it can be used to determine a protein’s mass and identity.
Mass spectrometry is a technique for analyzing ionized forms of molecules in the gas phase. It is most readily applied to gases or to volatile liquids that easily release gas-
Protein MassTwo widely used methods, matrix-
After gas-
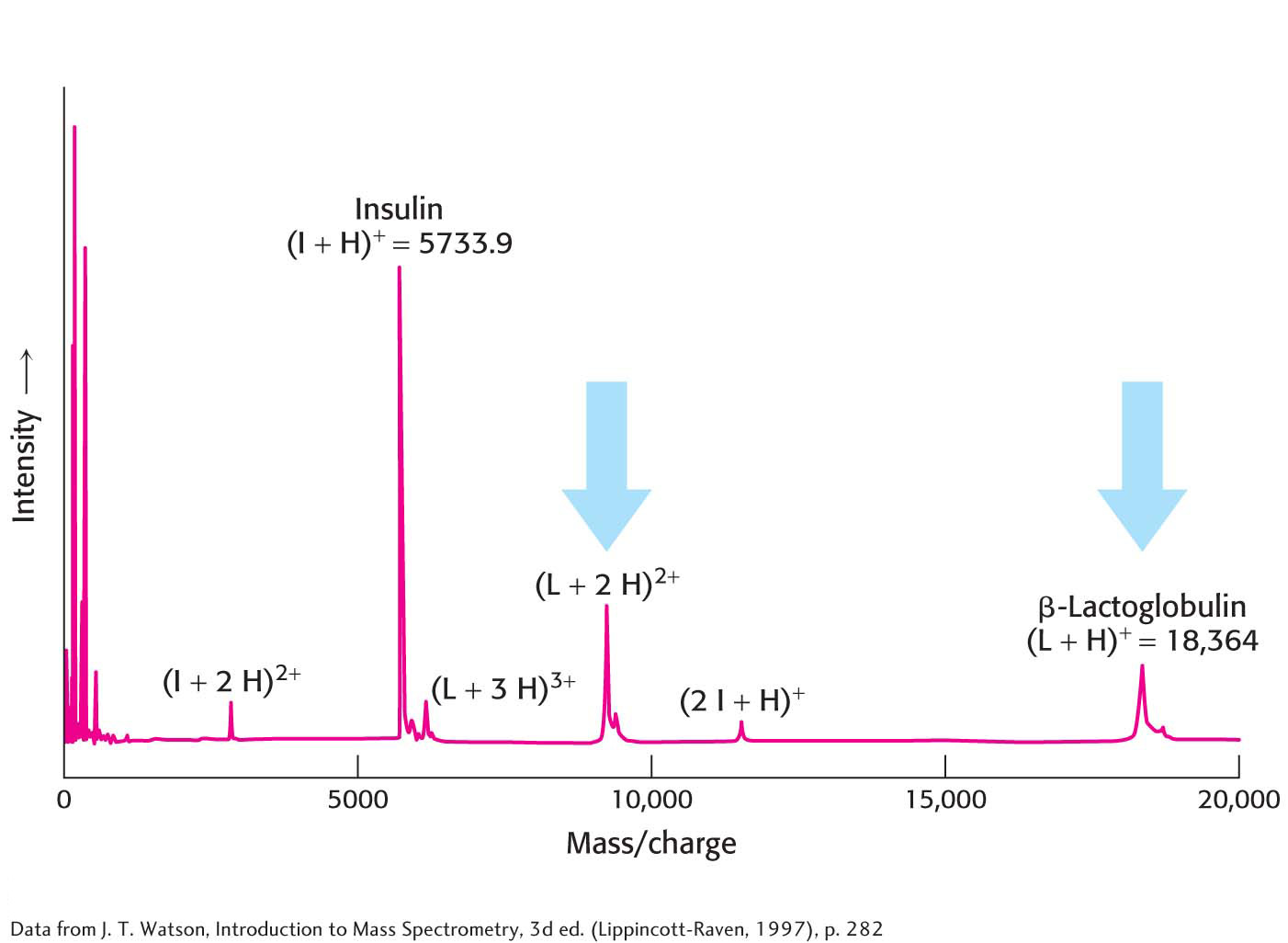
Protein IdentityAlthough protein masses serve as convenient name tags for distinguishing proteins, the mass of a given protein is usually not enough to uniquely identify it among all possible proteins within a cell. However, the mass of the parent protein along with the masses of several protein fragments produced by a specific cleavage method can provide unique identification. Suppose we wish to identify proteins within a two-
Protein SequenceHow can we employ mass spectrometry to sequence a protein? The use of mass spectrometry for protein sequencing takes advantage of the fact that ions of proteins that have been analyzed by a mass spectrometer, the precursor ions, can be broken into smaller peptide chains by bombardment with atoms of an inert gas such as helium or argon. These new fragments, or product ions, can be passed through a second mass analyzer for further mass characterization. The utilization of two mass analyzers arranged in this manner is referred to as tandem mass spectrometry. Importantly, the product-
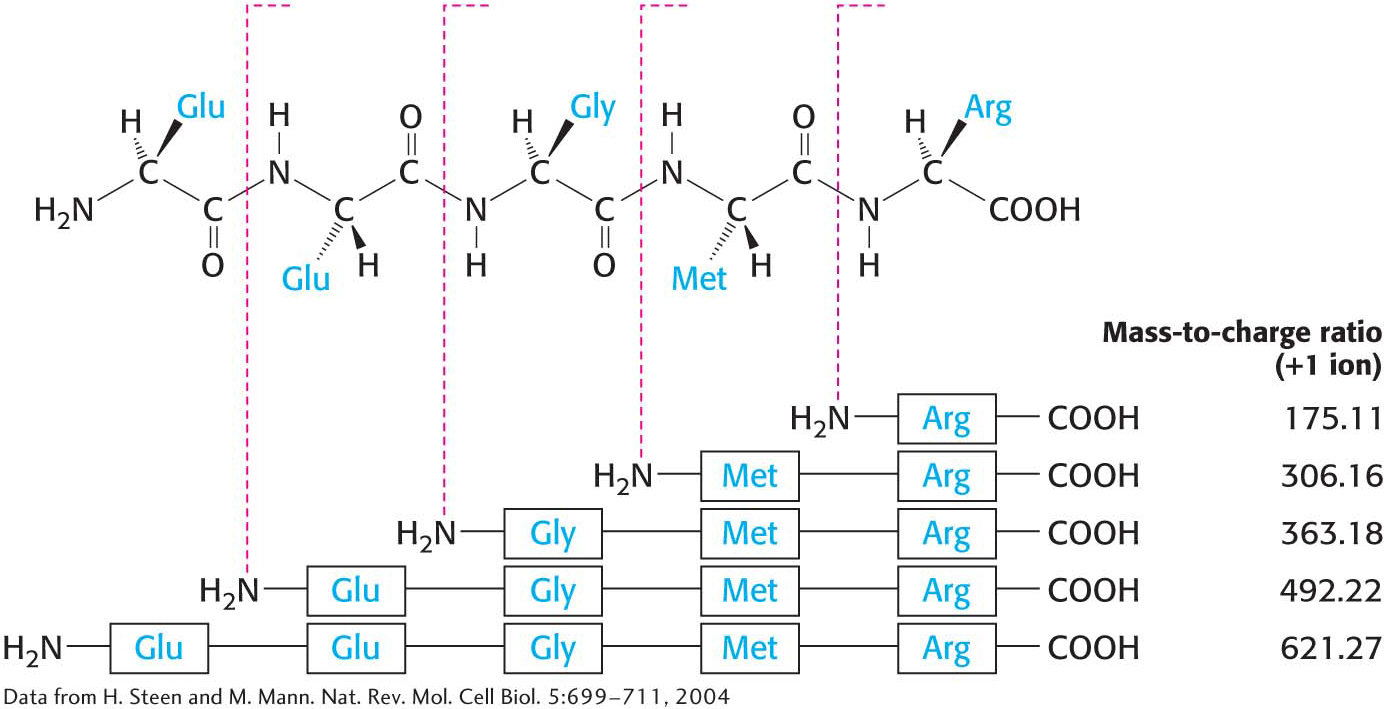
Amino Acid Sequences Are Sources of Many Kinds of Insight
QUICK QUIZ 4
Differentiate between amino acid composition and amino acid sequence.
Amino acid composition is simply the amino acids that are present in the protein. Many proteins can have the same amino acid composition. Amino acid sequence is the sequence of amino acids, or the primary structure, of the protein. Each protein has a unique amino acid sequence.
A protein’s amino acid sequence is a valuable source of insight into the protein’s function, structure, and history.
The sequence of a protein of interest can be compared with all other known sequences to ascertain whether significant similarities exist. Does this protein belong to an established family? A search for kinship between a newly sequenced protein and the millions of previously sequenced ones takes only a few seconds on a computer. If the newly isolated protein is a member of an established family of proteins, we can infer information about the protein’s structure and function. For instance, chymotrypsin and trypsin are members of the serine protease family, a clan of proteolytic enzymes that have a common catalytic mechanism based on a reactive serine residue. If the sequence of the newly isolated protein shows sequence similarity with trypsin or chymotrypsin, the result suggests that it, too, may be a serine protease.
Page 91Comparison of sequences of the same protein in different species yields a wealth of information about evolutionary pathways. Genealogical relations between species can be established from sequence differences between their proteins. We can even estimate the time at which two evolutionary lines diverged, thanks to the clocklike nature of random mutations. For example, a comparison of serum albumins found in primates indicates that human beings and African apes diverged 5 million years ago, not 30 million years ago as was once thought. Sequence analyses have opened a new perspective on the fossil record and the pathway of human evolution.
Amino acid sequences can be searched for the presence of internal repeats. Such internal repeats can reveal the history of an individual protein itself. Many proteins apparently have arisen by the duplication of primordial genes. For example, calmodulin, a ubiquitous calcium sensor in eukaryotes (Chapter 13), contains four similar calcium-
binding modules that arose by gene duplication (Figure 5.31). 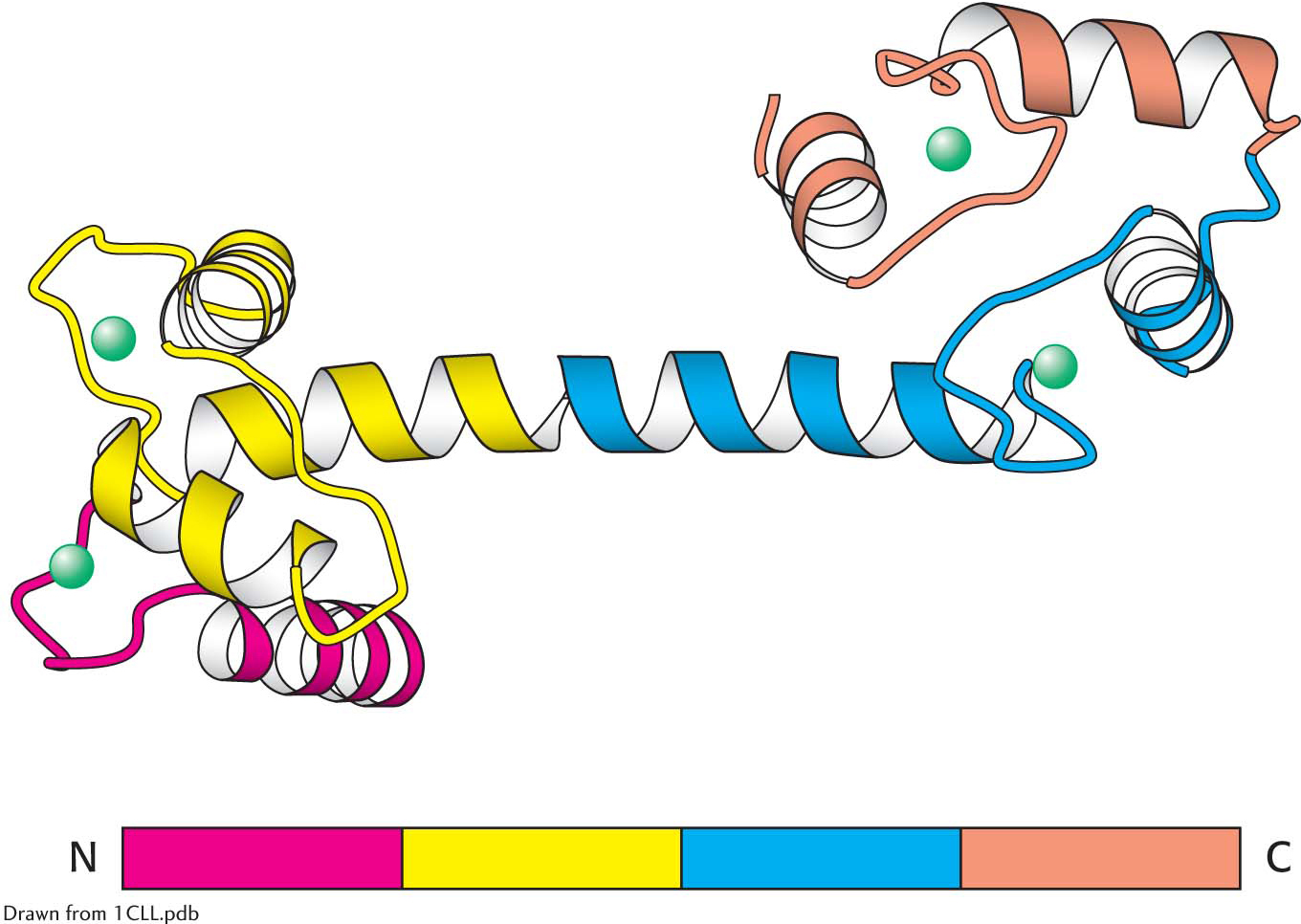 Figure 5.31 Repeating motifs in a protein chain. Calmodulin, a calcium sensor, contains four similar units (shown in red, yellow, blue, and orange) in a single polypeptide chain. Notice that each unit binds a calcium ion (shown in green).
Figure 5.31 Repeating motifs in a protein chain. Calmodulin, a calcium sensor, contains four similar units (shown in red, yellow, blue, and orange) in a single polypeptide chain. Notice that each unit binds a calcium ion (shown in green).Many proteins contain amino acid sequences that serve as signals designating their destinations or controlling their processing. For example, a protein destined for export from a cell or for location in a membrane contains a signal sequence, a stretch of about 20 hydrophobic residues near the amino terminus that directs the protein to the appropriate membrane (Chapter 40). Another protein may contain a stretch of amino acids that functions as a nuclear localization signal, directing the protein to the nucleus.
Sequence data allow a molecular understanding of diseases. Many diseases are caused by mutations in DNA that result in alterations in the amino acid sequence of a particular protein. These alterations often compromise the protein’s function. For instance, sickle-
cell anemia is caused by a change in a single amino acid in the primary structure of the β chain of hemoglobin (Chapter 9). Approximately 70% of the cases of cystic fibrosis are caused by the deletion of one particular amino acid out of the 1480 amino acids in the protein that controls chloride transport across cell membranes. Indeed, a major goal of biochemistry is to elucidate the molecular basis of disease with the hope that this understanding will lead to effective treatment. Protein sequence is a guide to nucleic acid information. Knowledge of a protein’s primary structure allows access to genomic information. DNA sequences that correspond to a part of the amino acid sequence can be synthesized on the basis of the genetic code. These DNA sequences can be used as probes to isolate the gene encoding the protein or the DNA corresponding to the mRNA, called the cDNA or complementary DNA (Chapter 41).