5.3 Complete Genomes Have Been Sequenced and Analyzed
The methods just described are extremely effective for the isolation and characterization of fragments of DNA. However, the genomes of organisms ranging from viruses to human beings contain considerably longer sequences, arranged in very specific ways crucial for their integrated functions. Is it possible to sequence complete genomes and analyze them? For small genomes, this sequencing was accomplished soon after DNA-sequencing methods were developed. Sanger and his coworkers determined the complete sequence of the 5386 bases in the genome of the ϕX174 DNA virus in 1977, just a quarter century after Sanger’s pioneering elucidation of the amino acid sequence of a protein. This tour de force was followed several years later by the determination of the sequence of human mitochondrial DNA, a double-stranded circular DNA molecule containing 16,569 base pairs. It encodes 2 ribosomal RNAs, 22 transfer RNAs, and 13 proteins. Many other viral genomes were sequenced in subsequent years. However, the genomes of free-living organisms presented a great challenge because even the simplest comprises more than 1 million base pairs. Thus, sequencing projects require both rapid sequencing techniques and efficient methods for assembling many short stretches of 300 to 500 base pairs into a complete sequence.
The genomes of organisms ranging from bacteria to multicellular eukaryotes have been sequenced
With the development of automatic DNA sequencers based on fluorescent dideoxynucleotide chain terminators, high-volume, rapid DNA sequencing became a reality. The genome sequence of the bacterium Haemophilus influenzae was determined in 1995 by using a “shotgun” approach. The genomic DNA was sheared randomly into fragments that were then sequenced. Computer programs assembled the complete sequence by matching up overlapping regions between fragments. The H. influenzae genome comprises 1,830,137 base pairs and encodes approximately 1740 proteins (Figure 5.25). Using similar approaches, as well as more advanced methods described below, investigators have determined the sequences of more than 10,000 bacterial and archaeal species, including key model organisms such as E. coli, Salmonella typhimurium, and Archaeoglobus fulgidus, as well as pathogenic organisms such as Yersina pestis (bubonic plague) and Bacillus anthracis (anthrax).
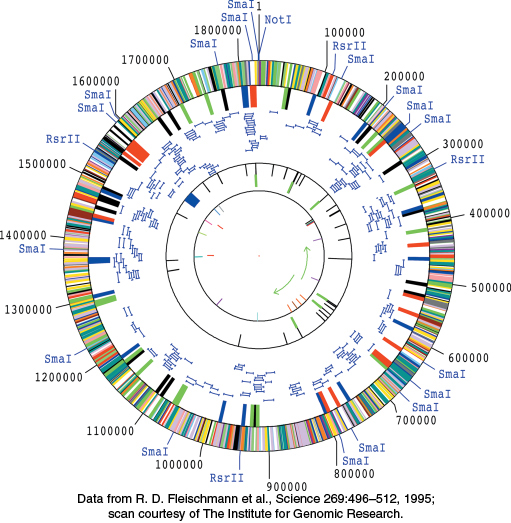
Figure 5.25: A complete genome. The diagram depicts the genome of Haemophilus influenzae, the first complete genome of a free-living organism to be sequenced. The genome encodes more than 1700 proteins and 70 RNA molecules. The likely function of approximately one-half of the proteins was determined by comparisons with sequences of proteins already characterized in other species.
[Data from R. D. Fleischmann et al., Science 269:496–512, 1995; scan courtesy of The Institute for Genomic Research.]
The first eukaryotic genome to be completely sequenced was that of baker’s yeast, Saccharomyces cerevisiae, in 1996. The yeast genome comprises approximately 12 million base pairs, distributed on 16 chromosomes, and encodes more than 6000 proteins. This achievement was followed in 1998 by the first complete sequencing of the genome of a multicellular organism, the nematode Caenorhabditis elegans, which contains 97 million base pairs. This genome includes more than 19,000 genes. The genomes of many additional organisms widely used in biological and biomedical research have now been sequenced, including those of the fruit fly Drosophila melanogaster, the model plant Arabidopsis thaliana, the mouse, the rat, and the dog. Note that even after a genome sequence has been considered complete, some sections, such as the repetitive sequences that make up heterochromatin, may be missing because these DNA sequences are very difficult to manipulate with the use of standard techniques.
The sequence of the human genome has been completed
The ultimate goal of much of genomics research has been the sequencing and analysis of the human genome. Given that the human genome comprises approximately 3 billion base pairs of DNA distributed among 24 chromosomes, the challenge of producing a complete sequence was daunting. However, through an organized international effort of academic laboratories and private companies, the human genome has progressed from a draft sequence first reported in 2001 to a finished sequence reported in late 2004 (Figure 5.26).
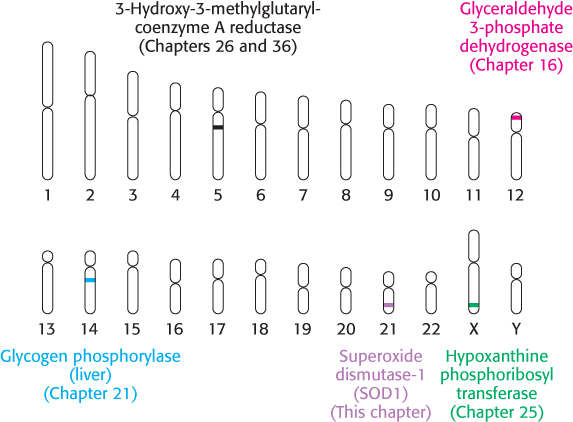
Figure 5.26: The human genome. The human genome is arrayed on 46 chromosomes—22 pairs of autosomes and the X and Y sex chromosomes. The locations of several genes associated with important pathways in biochemistry are highlighted.
The human genome is a rich source of information about many aspects of humanity, including biochemistry and evolution. Analysis of the genome will continue for many years to come. Developing an inventory of protein-encoding genes is one of the first tasks. At the beginning of the genome-sequencing project, the number of such genes was estimated to be approximately 100,000. With the availability of the completed (but not finished) genome, this estimate was reduced to between 30,000 and 35,000. With the finished sequence, the estimate fell to between 20,000 to 25,000. We will use the estimate of 23,000 throughout this book. The reduction in this estimate is due, in part, to the realization that there are a large number of pseudogenes, formerly functional genes that have accumulated mutations such that they no longer produce proteins. For example, more than half of the genomic regions that correspond to olfactory receptors—key molecules responsible for our sense of smell—are pseudogenes (Section 33.1). The corresponding regions in the genomes of other primates and rodents encode functional olfactory receptors. Nonetheless, the surprisingly small number of genes belies the complexity of the human proteome. Many genes encode more than one protein through mechanisms such as alternative splicing of mRNA and posttranslational modifications of proteins. The different proteins encoded by a single gene often display important variations in functional properties.
The human genome contains a large amount of DNA that does not encode proteins. A great challenge in modern biochemistry and genetics is to elucidate the roles of this noncoding DNA. Much of this DNA is present because of the existence of mobile genetic elements. These elements, related to retroviruses (Section 4.3), have inserted themselves throughout the genome over time. Most of these elements have accumulated mutations and are no longer functional. For example, more than 1 million Alu sequences, each approximately 300 bases in length, are present in the human genome. Alu sequences are examples of SINES, short interspersed elements. The human genome also includes nearly 1 million LINES, long interspersed elements, DNA sequences that can be as long as 10 kilobases (kb). The roles of these elements as neutral genetic parasites or instruments of genome evolution are under current investigation.
Next-generation sequencing methods enable the rapid determination of a complete genome sequence
Since the introduction of Sanger dideoxy method in the mid-1970s, significant advances have been made in DNA-sequencing technologies, enabling the readout of progressively longer sequences with higher fidelity and shorter run times. The development of next-generation sequencing (NGS) platforms has extended this capability to formerly unforeseen levels. By combining technological breakthroughs in the handling of very small amounts of liquid, high-resolution optics, and computing power, these methods have already made a significant impact on the ability to obtain whole genome sequences rapidly and cheaply (Chapter 1).
Next-generation sequencing refers to a family of technologies, each of which utilizes a unique approach for the determination of a DNA sequence. All of these methods are highly parallel: from 1 million to 1 billion DNA fragment sequences are acquired in a single experiment. How are NGS methods capable of attaining such a high number of parallel runs? Individual DNA fragments are amplified by PCR on a solid support—a single bead or a small region of a glass slide—such that clusters of identical DNA fragments are distinguishable by high-resolution imaging. These fragments then serve as templates for DNA polymerase, where the addition of nucleotide triphosphates is converted to a signal that can be detected in a highly sensitive manner. The technique used to detect individual base incorporation varies among the variety of NGS methods. However, most of these can be understood simply by considering the overall reaction of chain elongation catalyzed by DNA polymerase (Figure 5.27). In the reversible terminator method, the four nucleotides are added to the template DNA, with each base tagged with a unique fluorescent label and a reversibly-blocked 3´ end. The blocked end assures that only one phosphodiester linkage will form. Once the nucleotide is incorporated into the growing strand, it is identified by its fluorescent tag, the blocking agent is removed, and the process is repeated.
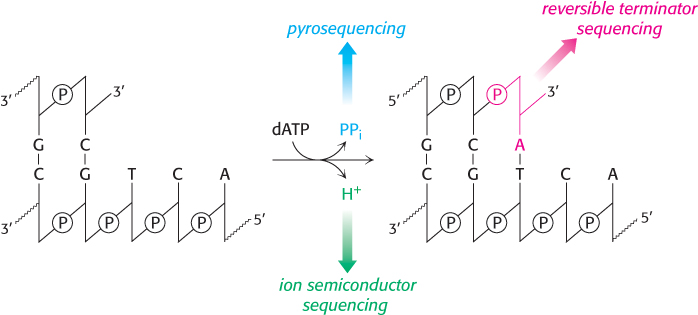
Figure 5.27: Detection methods in next-generation sequencing. Measurement of base incorporation in next-generation sequencing methods relies on the detection of the various products of the DNA polymerase reaction. Reversible terminator sequencing measures the nucleotide incorporation in a manner similar to Sanger sequencing, while pyrosequencing and ion semiconductor sequencing detect the release of pyrophosphate and protons, respectively.
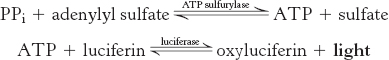
In pyrosequencing, nucleotides are added to the template DNA, one at a time in a defined order. One of the nucleotides will be incorporated into the growing strand, releasing a pyrophosphate which is detected by coupling the formation of pyrophosphate with the production of light by the sequential action of the enzymes ATP sulfurylase and luciferase:
The protocol for ion semiconductor sequencing is similar to pyrosequencing except that nucleotide incorporation is detected by sensitively measuring the very small changes in pH of the reaction mixture due to the release of proton upon nucleotide incorporation.
Regardless of the sequencing method, the technology exists to quantify the signal produced by millions of DNA fragment templates simultaneously. However, for many approaches, as few as 50 bases are read per fragment. Hence, significant computing power is required to both store the massive amounts of sequence data and perform the necessary alignments required to assemble a completed sequence. NGS methods are being used to answer an ever-growing number of questions in genomics, transcriptomics, and evolutionary biology, to name a few. Additionally, individual genome sequences will provide information about genetic variation within populations and may usher in an era of personalized medicine, when these data can be used to guide treatment decisions.
Comparative genomics has become a powerful research tool
Comparisons with genomes from other organisms are sources of insight into the human genome. The sequencing of the genome of the chimpanzee, our closest living relative, as well as that of other mammals that are widely used in biological research, such as the mouse and the rat, have been completed. Comparisons reveal that an astonishing 99% of human genes have counterparts in these rodent genomes. However, these genes have been substantially reassorted among chromosomes in the estimated 75 million years of evolution since humans and rodents had a common ancestor (Figure 5.28).
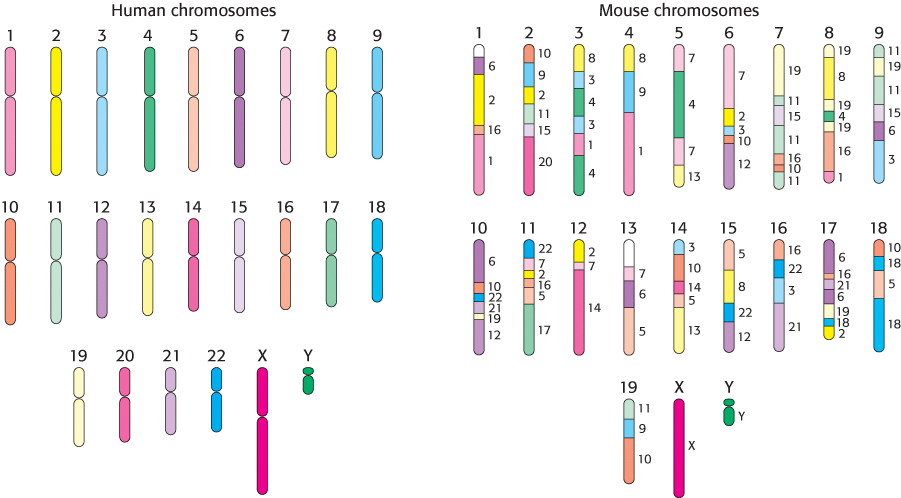
Figure 5.28: Genome comparison. A schematic comparison of the human genome and the mouse genome shows reassortment of large chromosomal fragments. The small numbers to the right of the mouse chromosomes indicate the human chromosome to which each region is most closely related.

A puffer fish.
[Beth Swanson/Shutterstock]
The genomes of other organisms also have been determined specifically for use in comparative genomics. For example, the genomes of two species of puffer fish, Takifugu rubripes and Tetraodon nigroviridis, have been determined. These genomes were selected because they are very small and lack much of the intergenic DNA present in such abundance in the human genome. The puffer fish genomes include fewer than 400 megabase pairs (Mbp), one-eighth of the number in the human genome, yet the puffer fish and human genomes contain essentially the same number of genes. Comparison of the genomes of these species with that of humans revealed more than 1000 formerly unrecognized human genes. Furthermore, comparison of the two species of puffer fish, which had a common ancestor approximately 25 million years ago, is a source of insight into more-recent events in evolution. Comparative genomics is a powerful tool, both for interpreting the human genome and for understanding major events in the origin of genera and species.