7.4 Mutations in Genes Encoding Hemoglobin Subunits Can Result in Disease
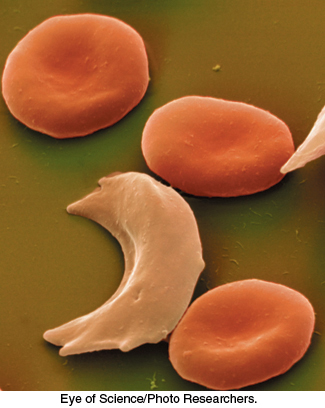
Figure 7.24: Sickled red blood cells. A micrograph showing a sickled red blood cell adjacent to normally shaped red blood cells.
[Eye of Science/Photo Researchers.]
In modern times, particularly after the sequencing of the human genome, it is routine to think of genetically encoded variations in protein sequence as a factor in specific diseases. The notion that diseases might be caused by molecular defects was proposed by Linus Pauling in 1949 (four years before Watson and Crick’s proposal of the DNA double helix) to explain the blood disease sickle-cell anemia. The name of the disorder comes from the abnormal sickle shape of red blood cells deprived of oxygen in people suffering from this disease (Figure 7.24). Pauling proposed that sickle-cell anemia might be caused by a specific variation in the amino acid sequence of one hemoglobin chain. Today, we know that this bold hypothesis is correct. In fact, approximately 7% of the world’s population are carriers of some disorder of hemoglobin caused by a variation in its amino acid sequence. In concluding this chapter, we will focus on the two most important of these disorders, sickle-cell anemia and thalassemia.
Sickle-cell anemia results from the aggregation of mutated deoxyhemoglobin molecules
 People with sickled red blood cells experience a number of dangerous symptoms. Examination of the contents of these red cells reveals that the hemoglobin molecules have formed large fibrous aggregates (Figure 7.25). These fibers extend across the red blood cells, distorting them so that they clog small capillaries and impair blood flow. In addition, red cells from sickle cell patients are more adherent to the walls of blood vessels than those from normal individuals, prolonging the opportunity for capillary occlusion. The results may be painful swelling of the extremities and a higher risk of stroke or bacterial infection (due to poor circulation). The sickled red cells also do not remain in circulation as long as normal cells do, leading to anemia.
People with sickled red blood cells experience a number of dangerous symptoms. Examination of the contents of these red cells reveals that the hemoglobin molecules have formed large fibrous aggregates (Figure 7.25). These fibers extend across the red blood cells, distorting them so that they clog small capillaries and impair blood flow. In addition, red cells from sickle cell patients are more adherent to the walls of blood vessels than those from normal individuals, prolonging the opportunity for capillary occlusion. The results may be painful swelling of the extremities and a higher risk of stroke or bacterial infection (due to poor circulation). The sickled red cells also do not remain in circulation as long as normal cells do, leading to anemia.
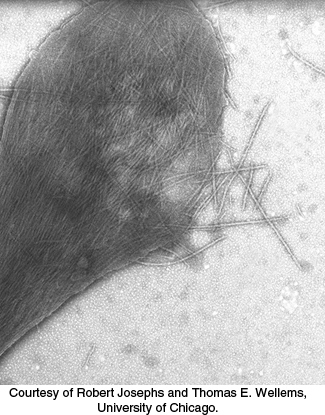
Figure 7.25: Sickle-cell hemoglobin fibers. An electron micrograph depicting a ruptured sickled red blood cell with fibers of sickle-cell hemoglobin emerging.
[Courtesy of Robert Josephs and Thomas E. Wellems, University of Chicago.]
What is the molecular defect associated with sickle-cell anemia? Vernon Ingram demonstrated in 1956 that a single amino acid substitution in the β chain of hemoglobin is responsible—namely, the replacement of a glutamate residue with valine in position 6. The mutated form is referred to as hemoglobin S (HbS). In people with sickle-cell anemia, both alleles of the hemoglobin β-chain gene (HbB) are mutated. The HbS substitution substantially decreases the solubility of deoxyhemoglobin, although it does not markedly alter the properties of oxyhemoglobin.
Examination of the structure of hemoglobin S reveals that the new valine residue lies on the surface of the T-state molecule (Figure 7.26). This new hydrophobic patch interacts with another hydrophobic patch formed by Phe 85 and Leu 88 of the β chain of a neighboring molecule to initiate the aggregation process. More-detailed analysis reveals that a single hemoglobin S fiber is formed from 14 chains of multiple interlinked hemoglobin molecules. Why do these aggregates not form when hemoglobin S is oxygenated? Oxyhemoglobin S is in the R state, and residues Phe 85 and Leu 88 on the β chain are largely buried inside the hemoglobin assembly. In the absence of a partner with which to interact, the surface Val residue in position 6 is benign (Figure 7.27).
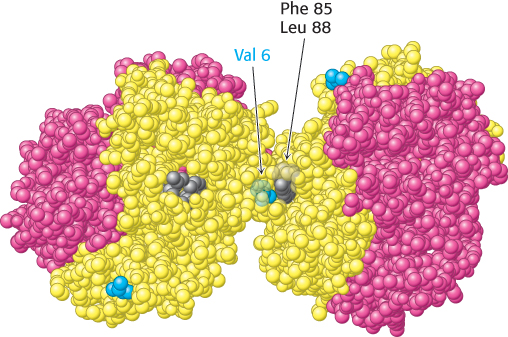
Figure 7.26: Deoxygenated hemoglobin S. The interaction between Val 6 (blue) on a β chain of one hemoglobin molecule and a hydrophobic patch formed by Phe 85 and Leu 88 (gray) on a β chain of another deoxygenated hemoglobin molecule leads to hemoglobin aggregation. The exposed Val 6 residues of other β chains participate in other such interactions in hemoglobin S fibers.
[Drawn from 2HBS.pdb.]
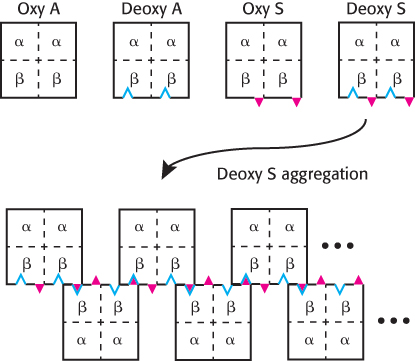
Figure 7.27: The formation of HbS aggregates. The mutation to Val 6 in hemoglobin S is represented by the red triangles, while the hydrophobic patch formed by Phe 85 and Leu 88 in deoxyhemoglobin is represented by the blue indentations. When HbS is in its deoxy form, it exhibits the complementary features necessary for aggregation.
Approximately 1 in 100 West Africans suffer from sickle-cell anemia. Given the often devastating consequences of the disease, why is the HbS mutation so prevalent in Africa and in some other regions? Recall that both copies of the HbB gene are mutated in people with sickle-cell anemia. Individuals with one copy of the HbB gene and one copy of the HbS gene are said to have sickle-cell trait because they can pass the HbS gene to their offspring. While sickle-cell trait is considered a benign condition, rare complications have been identified, including an increased risk of exercise-related death in high-performance athletes. However, people with sickle-cell trait exhibit enhanced resistance to malaria, a disease carried by a parasite, Plasmodium falciparum, that lives within red blood cells at one stage in its life cycle. The dire effect of malaria on health and reproductive likelihood in historically endemic regions has favored people with sickle-cell trait, increasing the prevalence of the HbS allele (Figure 7.28).
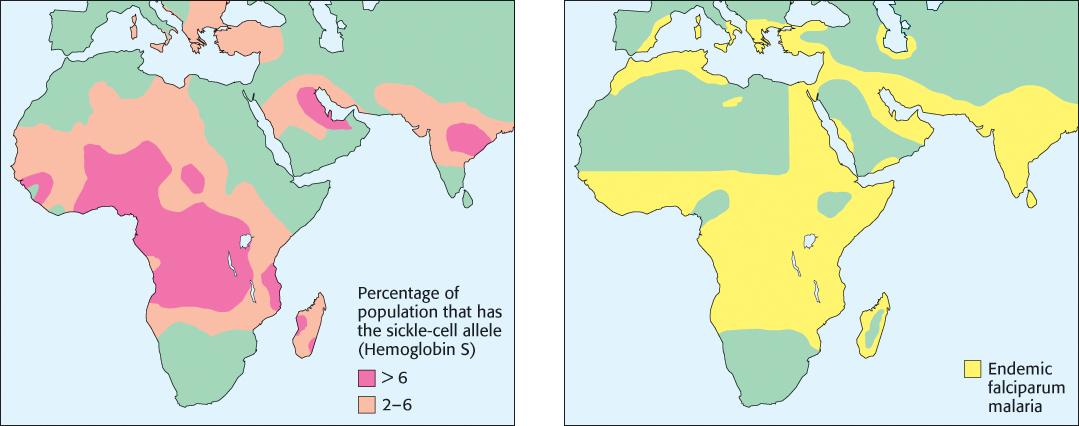
Figure 7.28: Sickle-cell trait and malaria. A significant correlation is observed between regions with a high frequency of the HbS allele and regions with a high prevalence of malaria.
Thalassemia is caused by an imbalanced production of hemoglobin chains
Sickle-cell anemia is caused by the substitution of a single specific amino acid in one hemoglobin chain. Thalassemia, the other prevalent inherited disorder of hemoglobin, is caused by the loss or substantial reduction of a single hemoglobin chain. The result is low levels of functional hemoglobin and a decreased production of red blood cells, which may lead to anemia, fatigue, pale skin, and spleen and liver malfunction. Thalassemia is a set of related diseases. In α-thalassemia, the α chain of hemoglobin is not produced in sufficient quantity. Consequently, hemoglobin tetramers form that contain only the β chain. These tetramers, referred to as hemoglobin H (HbH), bind oxygen with high affinity and no cooperativity. Thus, oxygen release in the tissues is poor. In β-thalassemia, the β chain of hemoglobin is not produced in sufficient quantity. In the absence of β chains, the α chains form insoluble aggregates that precipitate inside immature red blood cells. The loss of red blood cells results in anemia. The most severe form of β-thalassemia is called thalassemia major or Cooley anemia.
Both α- and β-thalassemia are associated with many different genetic variations and display a wide range of clinical severity. The most severe forms of α-thalassemia are usually fatal shortly before or just after birth. However, these forms are relatively rare. An examination of the repertoire of hemoglobin genes in the human genome provides one explanation. Normally, humans have not two but four alleles for the α chain, arranged such that the two genes are located adjacent to each other on one end of each chromosome 16. Thus, the complete loss of α-chain expression requires the disruption of four alleles. β-Thalassemia is more common because humans normally have only two alleles for the β chain, one on each copy of chromosome 11.
The accumulation of free alpha-hemoglobin chains is prevented
The presence of four genes expressing the α chain, compared with two for the β chain, suggests that the α chain would be produced in excess (given the overly simple assumption that protein expression from each gene is comparable). If this is correct, why doesn’t the excess α chain precipitate? One mechanism for maintaining α chains in solution was revealed by the discovery of an 11-kDa protein in red blood cells called α-hemoglobin stabilizing protein (AHSP). This protein forms a soluble complex specifically with newly synthesized α-chain monomers. The crystal structure of a complex between AHSP and α-hemoglobin reveals that AHSP binds to the same face of α-hemoglobin as does β-hemoglobin (Figure 7.29). AHSP binds the α chain in both the deoxygenated and oxygenated forms. In the complex with oxygen bound, the distal histidine, rather than the proximal histidine, binds the iron atom.
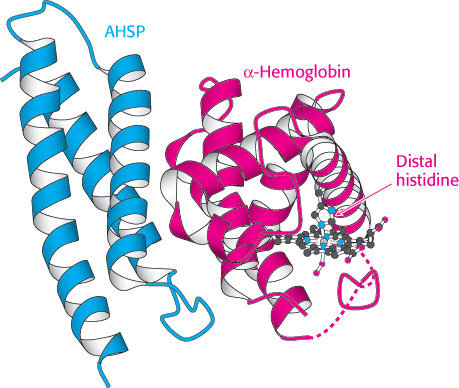
Figure 7.29:  Stabilizing free α-hemoglobin. The structure of a complex between AHSP and α-hemoglobin is shown. In this complex, the iron atom is bound to oxygen and to the distal histidine. Notice that AHSP binds to the same surface of α-hemoglobin as does β-hemoglobin.
Stabilizing free α-hemoglobin. The structure of a complex between AHSP and α-hemoglobin is shown. In this complex, the iron atom is bound to oxygen and to the distal histidine. Notice that AHSP binds to the same surface of α-hemoglobin as does β-hemoglobin.
[Drawn from 1Y01.pdb.]
AHSP serves to bind and ensure the proper folding of α-hemoglobin as it is produced. As β-hemoglobin is expressed, it displaces AHSP because the α-hemoglobin–β-hemoglobin dimer is more stable than the α-hemoglobin–AHSP complex. Thus, AHSP prevents the misfolding, accumulation, and precipitation of free α-hemoglobin. Studies are under way to determine if mutations in the gene encoding AHSP play a role in modulating the severity of β-thalassemia.
Additional globins are encoded in the human genome
 In addition to the gene for myoglobin, the two genes for α-hemoglobin, and the one for β-hemoglobin, the human haploid genome contains other globin genes. We have already encountered fetal hemoglobin, which contains the γ chain in place of the β chain. Several other genes encode other hemoglobin subunits that are expressed during development, including the δ chain, the ε chain, and the ζ chain.
In addition to the gene for myoglobin, the two genes for α-hemoglobin, and the one for β-hemoglobin, the human haploid genome contains other globin genes. We have already encountered fetal hemoglobin, which contains the γ chain in place of the β chain. Several other genes encode other hemoglobin subunits that are expressed during development, including the δ chain, the ε chain, and the ζ chain.
Examination of the human genome sequence has revealed two additional globins. Both of these proteins are monomeric proteins, more similar to myoglobin than to hemoglobin. The first, neuroglobin, is expressed primarily in the brain and at especially high levels in the retina. Neuroglobin may play a role in protecting neural tissues from hypoxia (insufficient oxygen). The second, cytoglobin, is expressed more widely throughout the body. Structural and spectroscopic studies reveal that, in both neuroglobin and cytoglobin, the proximal and the distal histidines are coordinated to the iron atom in the deoxy form. Oxygen binding displaces the distal histidine. Future studies should more completely elucidate the functions of these members of the globin family.