Chapter 23
1. When the proteins are denatured, all of the peptide bonds are accessible to proteolytic enzymes. If the three-
2. First, the ubiquitin-
3. (a) 7; (b) 4; (c) 2; (d) 10; (e) 5; (f ) 3; (g) 9; (h) 1; (i) 6; ( j) 8.
4. (a) The ATPase activity of the 26S proteasome resides in the 19S subunit. The energy of ATP hydrolysis is used to unfold the substrate, which is too large to enter the catalytic barrel. ATP may also be required for translocation of the substrate into the barrel.
(b) Substantiates the answer in part a. Because they are small, the peptides do not need to be unfolded. Moreover, small peptides could probably enter all at once and not require translocation.
5. (a) Pyruvate; (b) oxaloacetate; (c) α-ketoglutarate; (d) α-ketoisocaproate; (e) phenylpyruvate; (f ) hydroxyphenylpyruvate.
6. (a) Aspartate + α-ketoglutarate + GTP + ATP + 2 H2O + NADH + H+ → ½ glucose + glutamate + CO2 + ADP + GDP + NAD+ + 2 Pi.
The required coenzymes are pyridoxal phosphate in the transamination reaction and NAD+/NADH in the redox reactions.
(b) Aspartate + CO2 + NH4+ + 3 ATP + NAD+ + 4 H2O → oxaloacetate + urea + 2 ADP + 4 Pi + AMP + NADH + H+.
7. In the eukaryotic proteasome, the distinct β subunits have different substrate specificities, allowing proteins to be more thoroughly degraded.
8. The six subunits probably exist as a heterohexamer. Cross-
9. Thiamine pyrophosphate.
10. Aminotransferases transfer the α-amino group to α-ketoglutarate to form glutamate. Glutamate is oxidatively deaminated to form an ammonium ion.
11. Aspartate (oxaloacetate), glutamate (α-ketoglutarate), alanine (pyruvate).
12. Serine and threonine.
13. They are either fuels for the citric acid cycle, components of the citric acid cycle, or molecules that can be converted into a fuel for the citric acid cycle in one step.
A33
14. It acts as an electron sink.
15. Carbamoyl phosphate and aspartate.
16. (a) 4; (b) 5; (c) 1; (d) 6; (e) 7; (f ) 3; (g) 2.
17. A, arginine; B, citrulline; C, ornithine; D, arginosuccinate. The order of appearance: C, B, D, A.
18.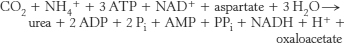 .
.
Four high-
19. The synthesis of fumarate by the urea cycle is important because it links the urea cycle and the citric acid cycle. Fumarate is hydrated to malate, which, in turn, is oxidized to oxaloacetate. Oxaloacetate has several possible fates: (1) transamination to aspartate, (2) conversion into glucose by the gluconeogenic pathway, (3) condensation with acetyl CoA to form citrate, or (4) conversion into pyruvate. You can collect.
20. Ornithine transcarbamoylase (Compound A is analogous to PALA; Chapter 10).
21. Ammonia could lead to the amination of α-ketoglutarate, producing a high concentration of glutamate in an unregulated fashion. α-Ketoglutarate for glutamate synthesis could be removed from the citric acid cycle, thereby diminishing the cell’s respiration capacity.
22. The mass spectrometric analysis strongly suggests that three enzymes—
23. Benzoate, phenylacetate, and arginine would be given to supplement a protein-
24. The liver is the primary tissue for capturing nitrogen as urea. If the liver is damaged (for instance, by hepatitis or the excessive consumption of alcohol), free ammonia is released into the blood.
25. This defect can be partly bypassed by providing a surplus of arginine in the diet and restricting the total protein intake. In the liver, arginine is split into urea and ornithine, which then reacts with carbamoyl phosphate to form citrulline. This urea-
26. Aspartame, a dipeptide ester (l-aspartyl-
27. N-Acetylglutamate is synthesized from acetyl CoA and glutamate. Once again, acetyl CoA serves as an activated acetyl donor. This reaction is catalyzed by N-acetylglutamate synthase.
28. Not all proteins are created equal: some are more important than others. Some proteins would be degraded to provide the missing amino acid. The nitrogen from the other amino acids would be excreted as urea. Consequently, more nitrogen would be excreted than ingested.
29. The carbon skeletons of ketogenic amino acids can be converted into ketone bodies or fatty acids. Only leucine and lysine are purely ketogenic. Glucogenic amino acids are those whose carbon skeletons can be converted into glucose.
30. As shown in Figure 23.28, alanine, a gluconeogenic amino acid, is released during the metabolism of tryptophan to acetyl CoA and acetoacetyl CoA.
31. The branched-
32. Pyruvate (glycolysis and gluconeogenesis), acetyl CoA (citric acid cycle and fatty acid synthesis), acetoacetyl CoA (ketone-
33.
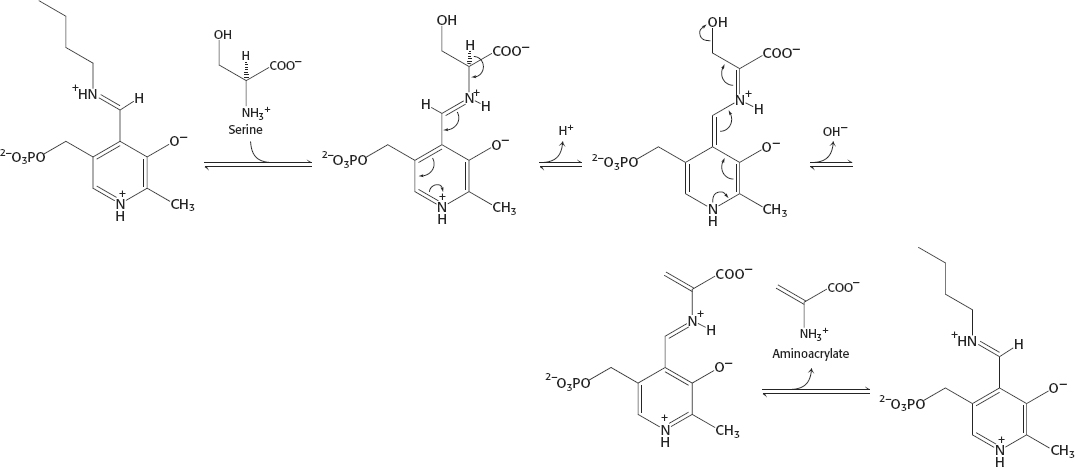
A34
34.
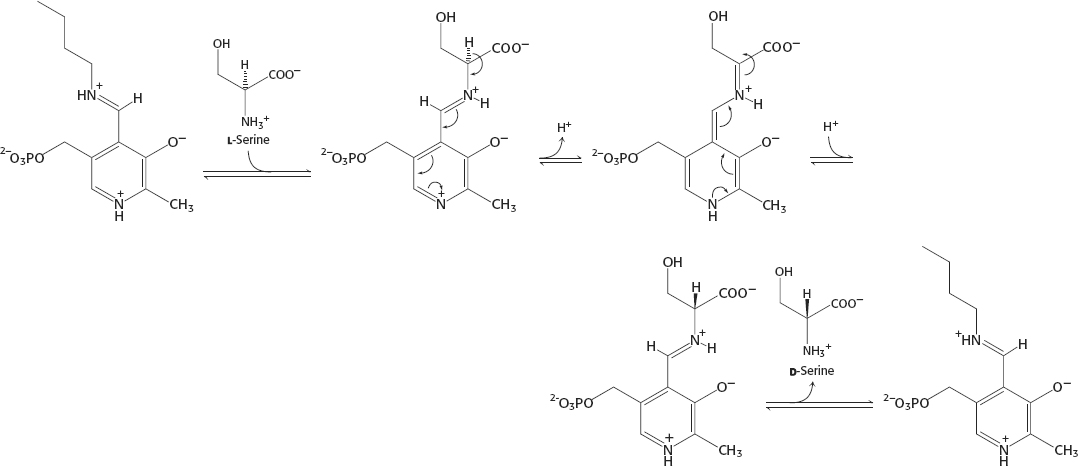
The equilibrium constant for the interconversion of l-serine and d-serine is exactly 1.
35. Double-
36. Exposure of such a domain suggests that a component of a multiprotein complex has failed to form properly or that one component has been synthesized in excess. This exposure leads to rapid degradation and the restoration of appropriate stoichiometries.
37. (a) Depletion of glycogen stores. When they are gone, proteins must be degraded to meet the glucose needs of the brain. The resulting amino acids are deaminated, and the nitrogen atoms are excreted as urea.
(b) The brain has adapted to the use of ketone bodies, which are derived from fatty acid catabolism. In other words, the brain is being powered by fatty acid breakdown.
(c) When the glycogen and lipid stores are gone, the only available energy source is protein.
38. The precise cause of all of the symptoms is not firmly established, but a likely explanation depends on the centrality of oxaloacetate to metabolism. A lack of pyruvate carboxylase would reduce the amount of oxaloacetate. The lack of oxaloacetate would reduce the activity of the citric acid cycle and so ATP would be generated by lactic acid formation. If the concentration of oxaloacetate is low, aspartate cannot be formed and the urea cycle would be compromised. Oxaloacetate is also required to form citrate, which transports acetyl CoA to the cytoplasm for fatty acid synthesis. Finally, oxaloacetate is required for gluconeogenesis.
39. Deamination to α-keto-
40. Glycogen phosphorylase. The coenzyme serves as an acid–
41. In the Cori cycle, the carbon atoms are transferred from muscle to liver as lactate. For lactate to be of any use, it must be reduced to pyruvate. This reduction requires high-
42. (a) Virtually no digestion in the absence of nucleotides.
(b) Protein digestion is greatly stimulated by the presence of ATP.
(c) AMP-
(d) The proteasome requires neither ATP nor PAN to digest small substrates.
(e) PAN and ATP hydrolysis may be required to unfold the peptide and translocate it into the proteasome.
(f) Although Thermoplasma PAN is not as effective with the other proteasomes, it nonetheless results in threefold to fourfold stimulation of digestion.
(g) In light of the fact that the archaea and eukarya diverged several billion years ago, the fact that Thermoplasma PAN can stimulate rabbit muscle suggests homology not only between the proteasomes, but also between PAN and the 19S subunit (most likely the ATPases) of the mammalian 26S proteasome.