14.1 Heterotrimeric G Proteins Transmit Signals and Reset Themselves
Epinephrine is a hormone secreted by the adrenal glands of mammals in response to internal and external stressors. It exerts a wide range of effects—referred to as the fight-or-flight response —to help organisms anticipate the need for rapid muscular activity, including acceleration of heart rate, dilation of the smooth muscle of the airways, and initiation of the breakdown of glycogen (Section 21.3) and fatty acids (Section 22.2). Epinephrine signaling begins with ligand binding to a protein called the β-adrenergic receptor (β-AR). The β-AR is a member of the largest class of cell-surface receptors, called the seven-transmembrane-helix (7TM) receptors. Members of this family are responsible for transmitting information initiated by signals as diverse as hormones, neurotransmitters, odorants, tastants, and even photons (Table 14.1). More than 20,000 such receptors are now known, including nearly 800 encoded in the human genome. Furthermore, about one-third of the marketed therapeutic drugs target receptors of this class. As the name indicates, these receptors contain seven helices that span the membrane bilayer (Figure 14.4).
|
|
|
|
|
|
|
|
|
|
Control of blood pressure |
|
|
Cell growth and differentiation |
|
|
|
|
|
|
|
|
|
|
Information from J. S. Gutkind, J. Biol. Chem. 273:1839–1842, 1998. |
Table 14.1: Biological functions mediated by 7TM receptors
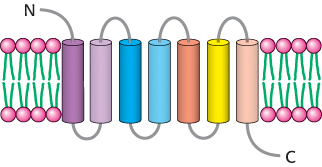
Figure 14.4: The 7TM receptor. Schematic representation of a 7TM receptor showing its passage through the membrane seven times.
The first member of the 7TM receptor family to have its three-dimensional structure determined was rhodopsin (Figure 14.5A), a protein in the retina of the eye that senses the presence of photons and initiates the signaling cascade responsible for visual sensation. A single lysine residue within rhodopsin is covalently modified by a form of vitamin A, 11-cis-retinal. This modification is located near the extracellular side of the receptor, within the region surrounded by the seven transmembrane helices. As will be considered in greater detail in Section 33.3, exposure to light induces the isomerization of 11-cis -retinal to its all-trans form, producing a structural change in the receptor that results in the initiation of an action potential that is ultimately interpreted by the brain as visual stimulus.
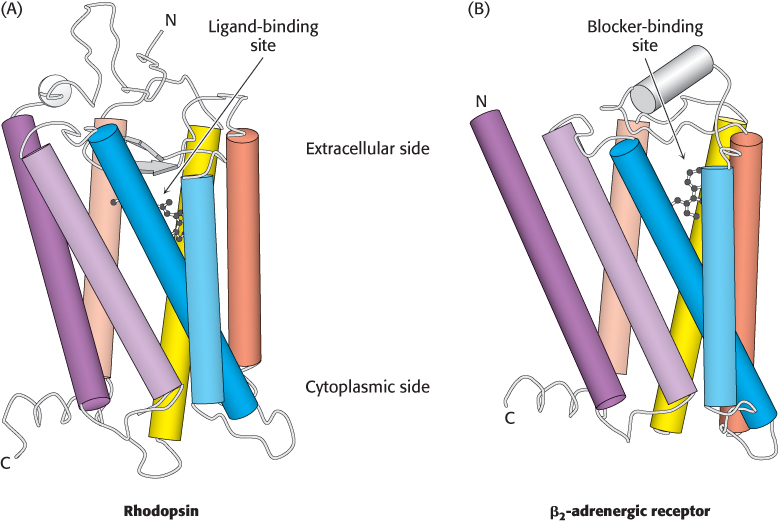
Figure 14.5: Structures of rhodopsin and the β2-adrenergic receptor. Three-dimensional structure of rhodopsin (A) and the β2-adrenergic receptor (β2-AR). (B). Notice the resemblance in the overall architecture of both receptors and the similar locations of the rhodopsin ligand 11-cis-retinal and the β2-AR blocker carazolol.
[Drawn from 1F88.pdb and 2RH1.pdb.]
In 2007, the first three-dimensional structure of the β2 subtype of the human adrenergic receptor (β2-AR) bound to an inhibitor was solved by x-ray crystallography. This inhibitor, carazolol, competes with epinephrine for binding to the β2-AR, much in the same way that competitive inhibitors act at enzyme active sites (Section 8.5).The structure of the β2-AR revealed considerable similarities with that of rhodopsin, particularly with respect to the locations of 11- cis-retinal in rhodopsin and the binding site for carazolol (Figure 14.5B).
Ligand binding to 7TM receptors leads to the activation of heterotrimeric G proteins
What is the next step in the pathway? The conformational change in the receptor’s cytoplasmic domain activates a protein called a G protein, named for the fact that it binds guanyl nucleotides. The activated G protein stimulates the activity of adenylate cyclase, an enzyme that catalyzes the conversion of ATP into cAMP. The G protein and adenylate cyclase remain attached to the membrane, whereas cAMP, a second messenger, can travel throughout the cell carrying the signal originally brought by the binding of epinephrine. Figure 14.6 provides a broad overview of these steps.
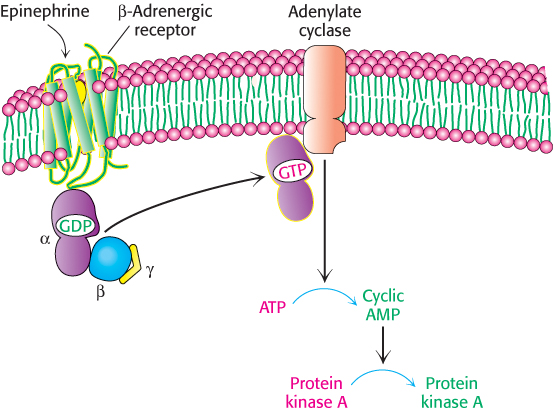
Figure 14.6: Activation of protein kinase A by a G-protein pathway. Hormone binding to a 7TM receptor initiates a signal-transduction pathway that acts through a G protein and cAMP to activate protein kinase A.
Let us consider the role of the G protein in this signaling pathway in greater detail. In its unactivated state, the G protein is bound to GDP. In this form, the G protein exists as a heterotrimer consisting of α, β, and γ subunits; the α subunit (referred to as Gα) binds the nucleotide (Figure 14.7). The α subunit is a member of the P-loop NTPase family (Section 9.4), and the P-loop participates in nucleotide binding. The α and γ subunits are usually anchored to the membrane by covalently attached fatty acids. The role of the hormone-bound receptor is to catalyze the exchange of GTP for bound GDP. The interaction between the hormone–β2-AR complex and the heterotrimeric G protein was illustrated in molecular detail when the crystal structure of this complex was determined in 2011. In this structure, a synthetic agonist, or small molecule that activates a receptor, was used to induce the active conformation of the β2-AR. Agonist binding results in the movement of two transmembrane helices, yielding an extensive interaction surface for the Gα subunit of the heterotrimer (Figure 14.8A). When bound to the receptor, the nucleotide-binding site of Gα opens substantially, enabling the displacement of GDP by GTP (Figure 14.8B).
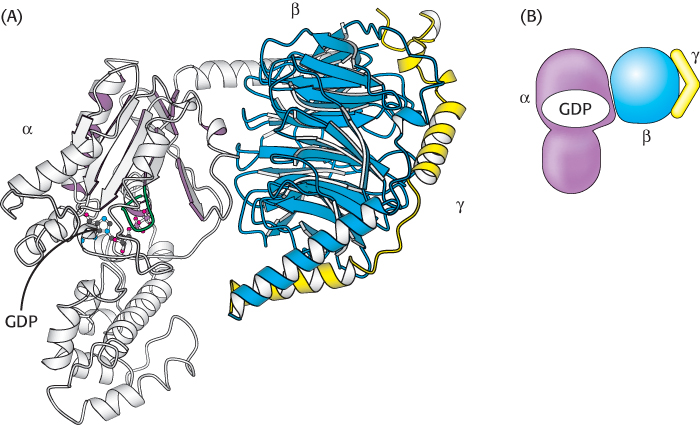
Figure 14.7: A heterotrimeric G protein. (A) A ribbon diagram shows the relation between the three subunits. In this complex, the α subunit (gray and purple) is bound to GDP. Notice that GDP is bound in a pocket close to the surface at which the α subunit interacts with the βγ dimer. (B) A schematic representation of the heterotrimeric G protein.
[Drawn from 1GOT.pdb.]
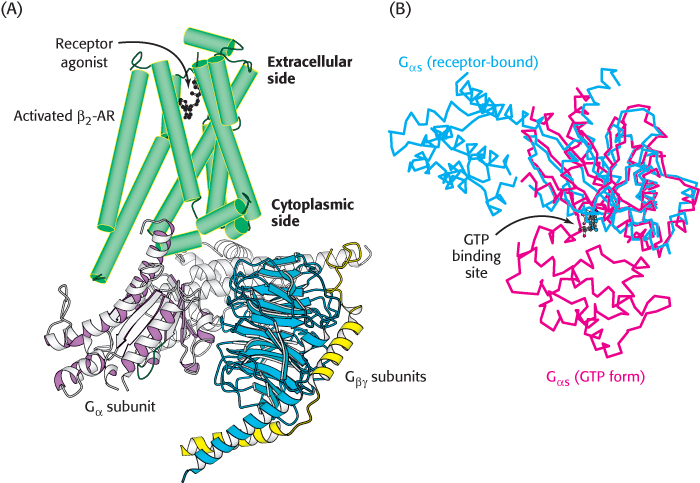
Figure 14.8: The complex between the activated β2-AR and a heterotrimeric G protein. (A) When the β2-AR (green) binds a receptor agonist, the cytoplasmic face of the receptor forms an interaction surface with the Gα subunit of a heterotrimeric G protein. (B) The interaction with the activated receptor leads to a substantial conformational change in the Gα protein, in which the GTP binding site is opened, enabling nucleotide exchange. In this figure, Gα in its GTP form is shown in red and in its receptor-bound form is shown in blue.
[Drawn from 3SN6.pdb and 1AZT.pdb.]
On GTP binding, the α subunit simultaneously dissociates from the βγ dimer (Gβγ), transmitting the signal that the receptor has bound its ligand. A single hormone–receptor complex can stimulate nucleotide exchange in many G-protein heterotrimers. Thus, hundreds of Gα molecules are converted from their GDP form into their GTP form for each bound molecule of hormone, giving an amplified response. Because they signal through G proteins, 7TM receptors are often called G-protein-coupled receptors (GPCRs).
Activated G proteins transmit signals by binding to other proteins
In the GTP form, the surface of Gα that had been bound to Gβγ has changed its conformation from the GDP form so that it no longer has a high affinity for Gβγ. This surface is now exposed for binding to other proteins. In the β-AR pathway, the new binding partner is adenylate cyclase, the enzyme that converts ATP into cAMP. This enzyme is a membrane protein that contains 12 membrane-spanning helices; two large cytoplasmic domains form the catalytic part of the enzyme (Figure 14.9). The interaction of Gα with adenylate cyclase favors a more catalytically active conformation of the enzyme, thus stimulating cAMP production. Indeed, the Gα subunit that participates in the β-AR pathway is called Gαs (“s” stands for stimulatory). The net result is that the binding of epinephrine to the receptor on the cell surface increases the rate of cAMP production inside the cell. The generation of cAMP by adenylate cyclase provides a second level of amplification because each activated adenylate cyclase can convert many molecules of ATP into cAMP.
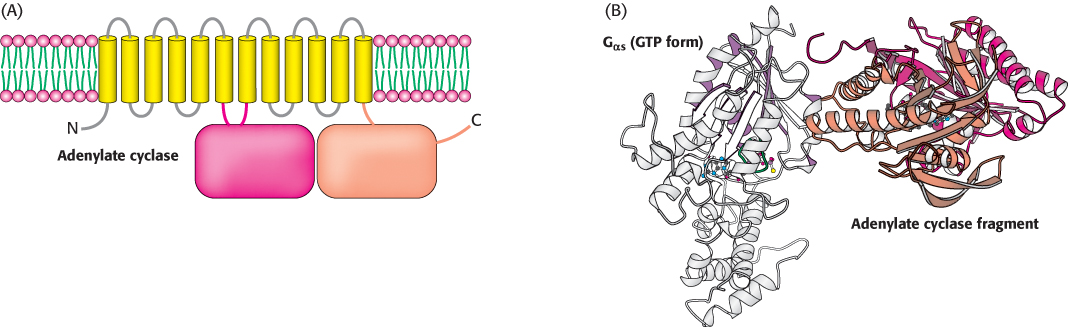
Figure 14.9:  Adenylate cyclase activation. (A) Adenylate cyclase is a membrane protein with two large intracellular domains that contain the catalytic apparatus. (B) The structure of a complex between Gα in its GTP form bound to a catalytic fragment from adenylate cyclase. Notice that the surface of Gα that had been bound to the βγ dimer now binds adenylate cyclase.
Adenylate cyclase activation. (A) Adenylate cyclase is a membrane protein with two large intracellular domains that contain the catalytic apparatus. (B) The structure of a complex between Gα in its GTP form bound to a catalytic fragment from adenylate cyclase. Notice that the surface of Gα that had been bound to the βγ dimer now binds adenylate cyclase.
[Drawn from 1AZS.pdb.]
Cyclic AMP stimulates the phosphorylation of many target proteins by activating protein kinase A
The increased concentration of cAMP can affect a wide range of cellular processes. In the muscle, cAMP stimulates the production of ATP for muscle contraction. In other cell types, cAMP enhances the degradation of storage fuels, increases the secretion of acid by the gastric mucosa, leads to the dispersion of melanin pigment granules, diminishes the aggregation of blood platelets, and induces the opening of chloride channels. How does cAMP influence so many cellular processes? Most effects of cAMP in eukaryotic cells are mediated by the activation of a single protein kinase. This key enzyme is protein kinase A (PKA).
As described earlier, PKA consists of two regulatory (R) chains and two catalytic (C) chains (R2C2; see Figure 10.16). In the absence of cAMP, the R2C2 complex is catalytically inactive. The binding of cAMP to the regulatory chains releases the catalytic chains, which are catalytically active on their own. Activated PKA then phosphorylates specific serine and threonine residues in many targets to alter their activity. For instance, PKA phosphorylates two enzymes that lead to the breakdown of glycogen, the polymeric store of glucose, and the inhibition of further glycogen synthesis (Section 21.3). PKA stimulates the expression of specific genes by phosphorylating a transcriptional activator called the cAMP response element binding (CREB) protein. This activity of PKA illustrates that signal-transduction pathways can extend into the nucleus to alter gene expression.
The signal-transduction pathway initiated by epinephrine is summarized in Figure 14.10.
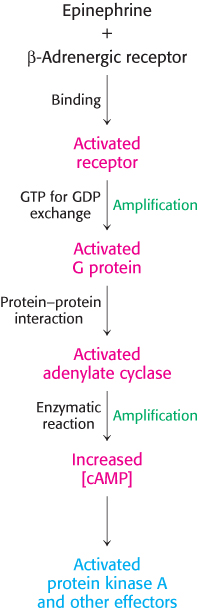
Figure 14.10: Epinephrine signaling pathway. The binding of epinephrine to the β-adrenergic receptor initiates the signal-transduction pathway. The process in each step is indicated (in black) at the left of each arrow. Steps that have the potential for signal amplification are indicated at the right in green.
G proteins spontaneously reset themselves through GTP hydrolysis
How is the signal initiated by epinephrine switched off? Gα subunits have intrinsic GTPase activity, which is used to hydrolyze bound GTP to GDP and Pi. This hydrolysis reaction is slow, however, requiring from seconds to minutes. Thus, the GTP form of Gα is able to activate downstream components of the signal-transduction pathway before it is deactivated by GTP hydrolysis. In essence, the bound GTP acts as a built-in clock that spontaneously resets the Gα subunit after a short time period. After GTP hydrolysis and the release of Pi, the GDP-bound form of Gα then reassociates with Gβγ to re-form the inactive heterotrimeric protein (Figure 14.11).
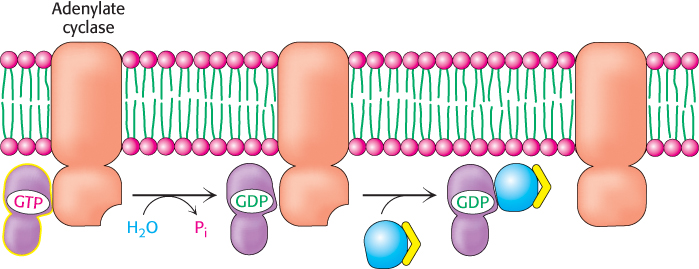
Figure 14.11: Resetting Gα. On hydrolysis of the bound GTP by the intrinsic GTPase activity of Gα, Gα reassociates with the βγ dimer to form the heterotrimeric G protein, thereby terminating the activation of adenylate cyclase.
The hormone-bound activated receptor must be reset as well to prevent the continuous activation of G proteins. This resetting is accomplished by two processes (Figure 14.12). First, the hormone dissociates, returning the receptor to its initial, unactivated state. The likelihood that the receptor remains in its unbound state depends on the extracellular concentration of hormone. Second, the signaling cascade initiated by the hormone–receptor complex activates a kinase that phosphorylates serine and threonine residues in the carboxyl-terminal tail of the receptor. These phosphorylation events result in the deactivation of the receptor. In the example under consideration, β-adrenergic-receptor kinase (also called G-protein receptor kinase 2, or GRK2) phosphorylates the carboxyl-terminal tail of the hormone–receptor complex but not the unoccupied receptor. Finally, the molecule β-arrestin binds to the phosphorylated receptor and further diminishes its ability to activate G proteins.
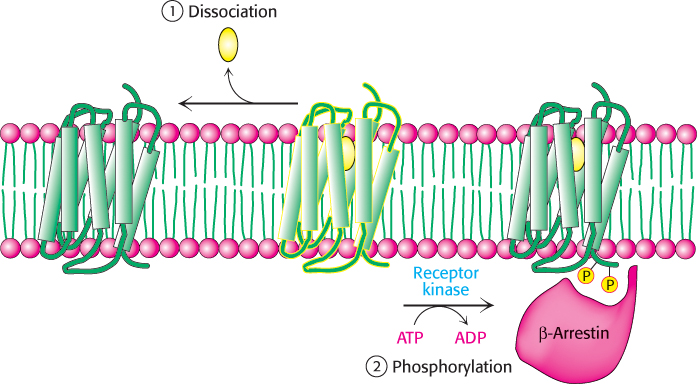
Figure 14.12: Signal termination. Signal transduction by the 7TM receptor is halted (1) by dissociation of the signal molecule from the receptor and (2) by phosphorylation of the cytoplasmic C-terminal tail of the receptor and the subsequent binding of β-arrestin.
Some 7TM receptors activate the phosphoinositide cascade
We now turn to another common second-messenger cascade, also employing a 7TM receptor, that is used by many hormones to evoke a variety of responses. The phosphoinositide cascade, like the cAMP cascade, converts extracellular signals into intracellular ones. The intracellular second messengers formed by activation of this pathway arise from the cleavage of phosphatidylinositol 4,5-bisphosphate (PIP2), a phospholipid present in cell membranes. An example of a signaling pathway based on the phosphoinositide cascade is the one triggered by the receptor for angiotensin II, a peptide hormone that controls blood pressure.
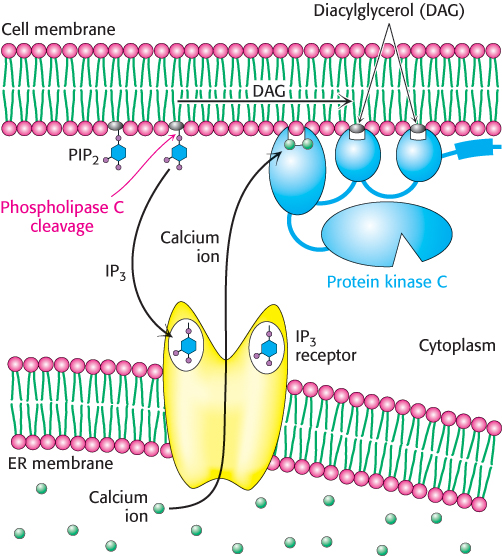
Figure 14.14: Phosphoinositide cascade. The cleavage of phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3) results in the release of calcium ions (owing to the opening of the IP3 receptor ion channels) and the activation of protein kinase C (owing to the binding of protein kinase C to free DAG in the membrane). Calcium ions bind to protein kinase C and help facilitate its activation.
Each type of 7TM receptor signals through a distinct G protein. Whereas the β-adrenergic receptor activates the G protein Gαs, the angiotensin II receptor activates a G protein called Gαq. In its GTP form, Gαq binds to and activates the β isoform of the enzyme phospholipase C. This enzyme catalyzes the cleavage of PIP2 into the two second messengers—inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG; Figure 14.13).
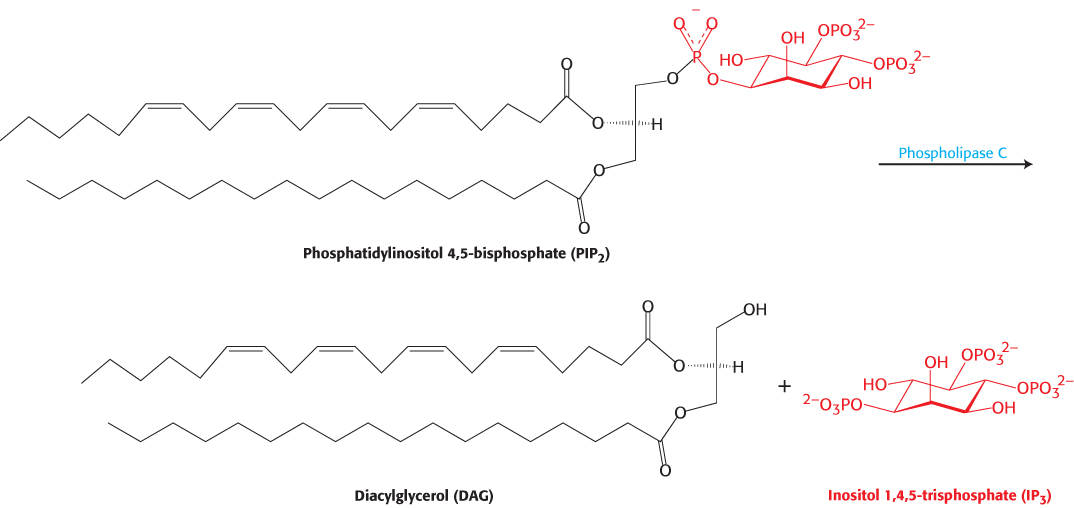
Figure 14.13: Phospholipase C reaction. Phospholipase C cleaves the membrane lipid phosphatidylinositol 4,5-bisphosphate into two second messengers: diacylglycerol, which remains in the membrane, and inositol 1,4,5-trisphosphate, which diffuses away from the membrane.
IP3 is soluble and diffuses away from the membrane. This second messenger causes the rapid release of Ca2+ from the intracellular stores in the endoplasmic reticulum (ER), which accumulates a reservoir of Ca2+ through the action of transporters such as Ca2+ ATPase (Section 13.2). On binding IP3, specific IP3-gated Ca2+– channel proteins in the ER membrane open to allow calcium ions to flow from the ER into the cytoplasm. Calcium ion is itself a signaling molecule: I t can bind proteins, including a ubiquitous signaling protein called calmodulin and enzymes such as protein kinase C. By such means, the elevated level of cytoplasmic Ca2+ triggers processes such as smooth-muscle contraction, glycogen breakdown, and vesicle release.
DAG remains in the plasma membrane. There, it activates protein kinase C (PKC), a protein kinase that phosphorylates serine and threonine residues in many target proteins. To bind DAG, the specialized DAG-binding domains of this kinase require bound calcium. Note that DAG and IP3 work in tandem: IP3 increases the Ca2+ concentration, and Ca2+ facilitates the DAG-mediated activation of protein kinase C. The phosphoinositide cascade is summarized in Figure 14.14. Both IP3 and DAG act transiently because they are converted into other species by phosphorylation or other processes.
Calcium ion is a widely used second messenger
Calcium ion participates in many signaling processes in addition to the phosphoinositide cascade. Several properties of this ion account for its widespread use as an intracellular messenger. First, fleeting changes in Ca2+ concentration are readily detected. At steady state, intracellular levels of Ca2+ must be kept low to prevent the precipitation of carboxylated and phosphorylated compounds, which form poorly soluble salts with Ca2+. Transport systems extrude Ca2+ from the cytoplasm, maintaining the cytoplasmic concentration of Ca2+ at approximately 100 nM, several orders of magnitude lower than that of the extracellular medium (Section 13.2). Given this low steady-state level, transient increases in Ca2+ concentration produced by signaling events can be readily sensed.
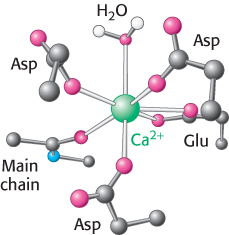
Figure 14.15: Calcium-binding site. In one common mode of binding, calcium is coordinated to six oxygen atoms of a protein and one (top) of water.
A second property of Ca2+ that makes it a highly suitable intracellular messenger is that it can bind tightly to proteins and induce substantial structural rearrangements. Calcium ions bind well to negatively charged oxygen atoms (from the side chains of glutamate and aspartate) and uncharged oxygen atoms (main-chain carbonyl groups and side-chain oxygen atoms from glutamine and asparagine; Figure 14.15). The capacity of Ca2+ to be coordinated to multiple ligands—from six to eight oxygen atoms—enables it to cross-link different segments of a protein and induce significant conformational changes.
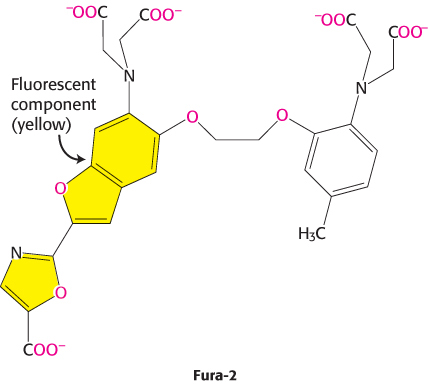
Our understanding of the role of Ca2+ in cellular processes has been greatly enhanced by our ability to detect changes in Ca2+ concentrations inside cells and even monitor these changes in real time. This ability depends on the use of specially designed dyes such as Fura-2 that bind Ca2+ and change their fluorescent properties on Ca2+ binding. Fura-2 binds Ca2+ through appropriately positioned oxygen atoms (shown in red) within its structure.
When such a dye is introduced into cells, changes in available Ca2+ concentration can be monitored with microscopes capable of detecting changes in fluorescence (Figure 14.16). Probes for sensing other second messengers such as cAMP also have been developed. These molecular-imaging agents are greatly enhancing our understanding of signal-transduction processes.
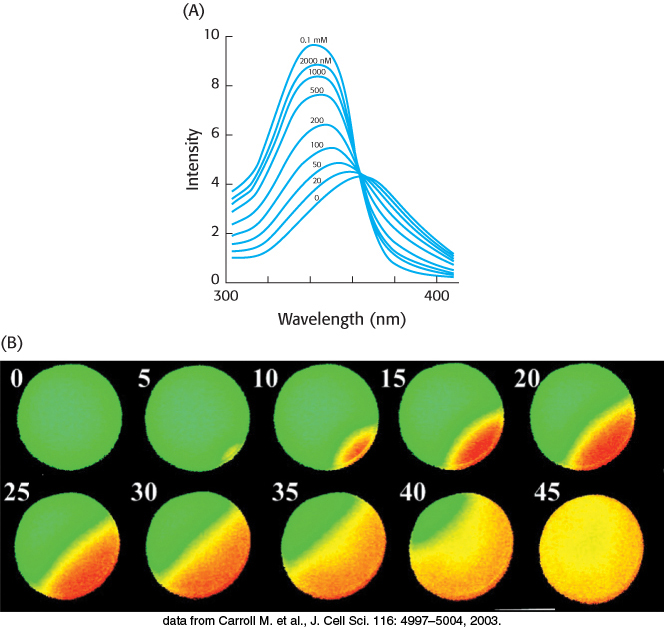
Figure 14.16: Calcium imaging. (A) The fluorescence spectra of the calcium-binding dye Fura-2 can be used to measure available calcium ion concentrations in solution and in cells. (B) A series of images show Ca2+ spreading across an egg cell following fertilization by sperm. These images were obtained through the use of Fura-2. The images are false colored: orange represents high Ca2+ concentrations, and green represents low Ca2+ concentrations.
[(A) Information from S. J. Lippard and J. M. Berg, Principles of Bioinorganic Chemistry (University Science Books, 1994), p. 193; (B) data from Carroll M. et al., J. Cell Sci. 116: 4997–5004, 2003.]
Calcium ion often activates the regulatory protein calmodulin
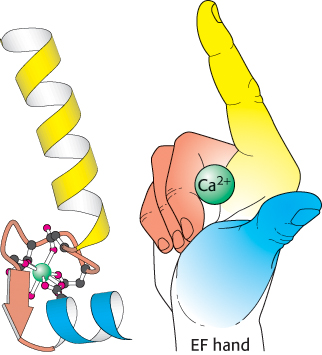
Figure 14.17:
EF hand. Formed by a helix-loop-helix unit, an EF hand is a binding site for Ca2+ in many calcium-sensing proteins. Here, the E helix is yellow, the F helix is blue, and calcium is represented by the green sphere. Note that the calcium ion is bound in a loop connecting two nearly perpendicular helices.
[Drawn from 1CLL.pdb.]
Calmodulin (CaM), a 17-kDa protein with four Ca2+ -binding sites, serves as a calcium sensor in nearly all eukaryotic cells. At cytoplasmic concentrations above about 500 nM, Ca2+ binds to and activates calmodulin. Calmodulin is a member of the EF-hand protein family. The EF hand is a Ca2+-binding motif that consists of a helix, a loop, and a second helix. This motif, originally discovered in the protein parvalbumin, was named the EF hand because the two key helices designated E and F in parvalbumin are positioned like the forefinger and thumb of the right hand (Figure 14.17). These two helices and the intervening loop form the Ca2+-binding motif. Seven oxygen atoms are coordinated to each Ca2+, six from the protein and one from a bound water molecule. Calmodulin is made up of four EF-hand motifs, each of which can bind a single Ca2+ ion.
The binding of Ca2+ to calmodulin induces substantial conformational changes in its EF hands, exposing hydrophobic surfaces that can be used to bind other proteins. Using its two sets of two EF hands, calmodulin clamps down around specific regions of target proteins, usually exposed α helices with appropriately positioned hydrophobic and charged groups (Figure 14.18). The Ca2+ –calmodulin complex stimulates a wide variety of enzymes, pumps, and other target proteins by inducing structural rearrangements in these binding partners. An especially noteworthy set of targets are several calmodulin-dependent protein kinases (CaM kinases) that phosphorylate many different proteins and regulate fuel metabolism, ionic permeability, neurotransmitter synthesis, and neurotransmitter release. We see here a recurring theme in signal-transduction pathways: the concentration of a second messenger is increased (in this case, Ca2+); the signal is sensed by a second-messenger-binding protein (in this case, calmodulin); and the second-messenger-binding protein acts to generate changes in enzymes (in this case, calmodulin-dependent kinases) that control effectors.
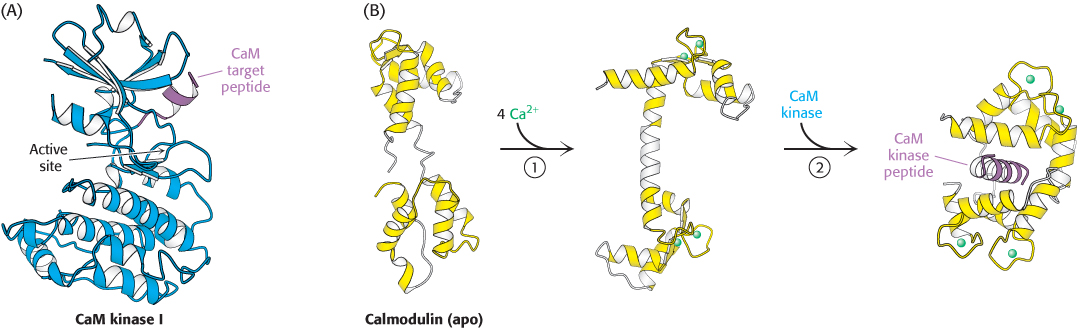
Figure 14.18:
Calmodulin binds to α helices. (A) An α helix (purple) in CaM kinase I is a target for calmodulin. (B) On Ca2+ binding to the apo, or calcium-free, form of calmodulin (1), the two halves of calmodulin clamp down around the target helix (2), binding it through hydrophobic and ionic interactions. In CaM kinase I, this interaction allows the enzyme to adopt an active conformation.
[Drawn from 1A06, 1CFD, 1CLL, and 1CM1.pdb.]