23.3 The First Step in Amino Acid Degradation Is the Removal of Nitrogen
What is the fate of amino acids released on protein digestion or turnover? The first call is for use as building blocks for biosynthetic reactions. However, any not needed as building blocks are degraded to compounds able to enter the metabolic mainstream. The amino group is first removed, and then the remaining carbon skeleton is metabolized to glucose, one of several citric acid cycle intermediates, or to acetyl CoA. The major site of amino acid degradation in mammals is the liver, although muscles readily degrade the branched-
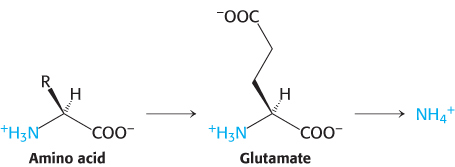
Alpha-amino groups are converted into ammonium ions by the oxidative deamination of glutamate
The α-amino group of many amino acids is transferred to α-ketoglutarate to form glutamate, which is then oxidatively deaminated to yield ammonium ion (NH4+).
688
Aminotransferases catalyze the transfer of an α-amino group from an α-amino acid to an α-ketoacid. These enzymes, also called transaminases, generally funnel α-amino groups from a variety of amino acids to α-ketoglutarate for conversion into NH4+.

Aspartate aminotransferase, one of the most important of these enzymes, catalyzes the transfer of the amino group of aspartate to α-ketoglutarate.

Alanine aminotransferase catalyzes the transfer of the amino group of alanine to α-ketoglutarate.

Transamination reactions are reversible and can thus be used to synthesize amino acids from α-ketoacids, as we shall see in Chapter 24.
The nitrogen atom in glutamate is converted into free ammonium ion by oxidative deamination, a reaction catalyzed by glutamate dehydrogenase. This enzyme is unusual in being able to utilize either NAD+ or NADP+, at least in some species. The reaction proceeds by dehydrogenation of the C—
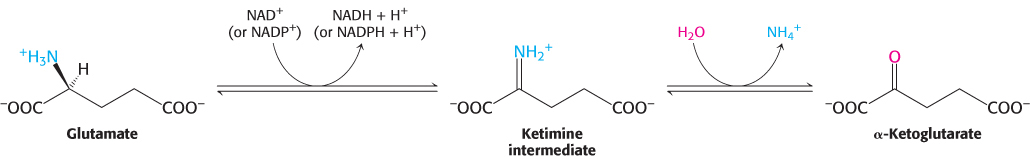
This reaction equilibrium constant is close to 1 in the liver, so the direction of the reaction is determined by the concentrations of reactants and products. Normally, the reaction is driven forward by the rapid removal of ammonium ion. Glutamate dehydrogenase, essentially a liver-
In mammals, but not in other organisms, glutamate dehydrogenase is allosterically inhibited by GTP and stimulated by ADP. These nucleotides exert their regulatory effects in a unique manner. An abortive complex is formed on the enzyme when a product is replaced by substrate before the reaction is complete. For instance, the enzyme bound to glutamate and NAD(P)H is an abortive complex. GTP facilitates the formation of such complexes while ADP destabilizes the complexes.
The sum of the reactions catalyzed by aminotransferases and glutamate dehydrogenase is
689
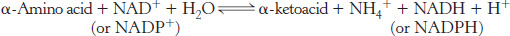
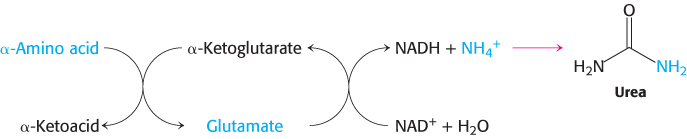
In most terrestrial vertebrates, NH4+ is converted into urea, which is excreted.
Mechanism: Pyridoxal phosphate forms Schiff-base intermediates in aminotransferases
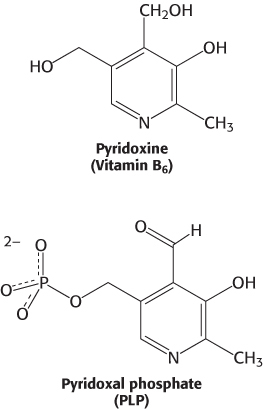
All aminotransferases contain the prosthetic group pyridoxal phosphate (PLP), which is derived from pyridoxine (vitamin B6). Pyridoxal phosphate includes a pyridine ring that is slightly basic to which is attached an OH group that is slightly acidic. Thus, pyridoxal phosphate derivatives can form a stable tautomeric form in which the pyridine nitrogen atom is protonated and, hence, positively charged, while the OH group loses a proton and hence is negatively charged, forming a phenolate.
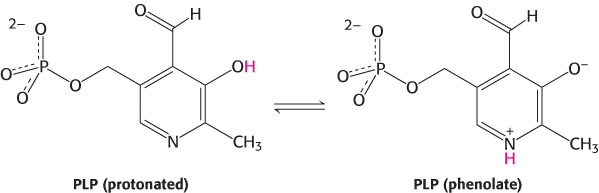
The most important functional group on PLP is the aldehyde. This group forms covalent Schiff-
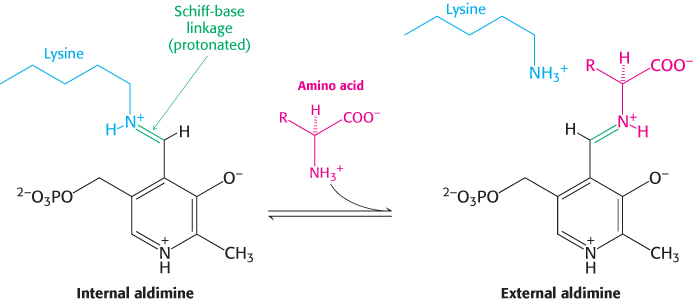
The α-amino group of the amino acid substrate displaces the ε-amino group of the active-
690
The Schiff base between the amino acid substrate and PLP, the external aldimine, loses a proton from the α-carbon atom of the amino acid to form a quinonoid intermediate (Figure 23.9). Reprotonation of this intermediate at the aldehyde carbon atom yields a ketimine. The ketimine is then hydrolyzed to an α-ketoacid and pyridoxamine phosphate (PMP). These steps constitute half of the transamination reaction.

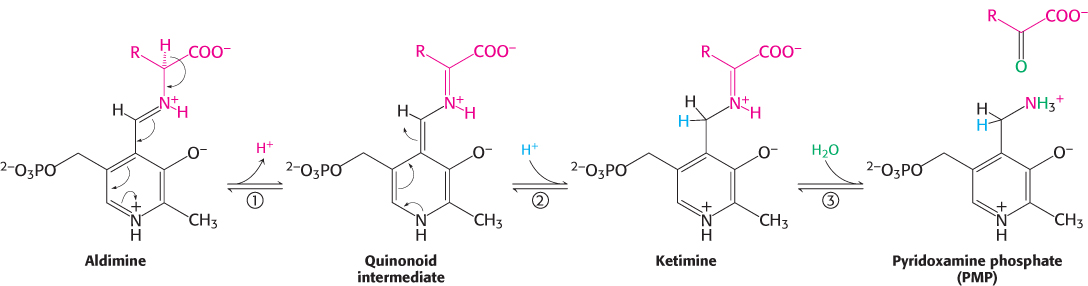
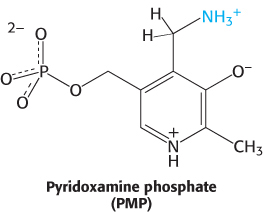
The second half takes place by the reverse of the preceding pathway. A second α-ketoacid reacts with the enzyme–

The sum of these partial reactions is

Aspartate aminotransferase is an archetypal pyridoxal-dependent transaminase
The mitochondrial enzyme aspartate aminotransferase provides an especially well studied example of PLP as a coenzyme for transamination reactions (Figure 23.10). X-
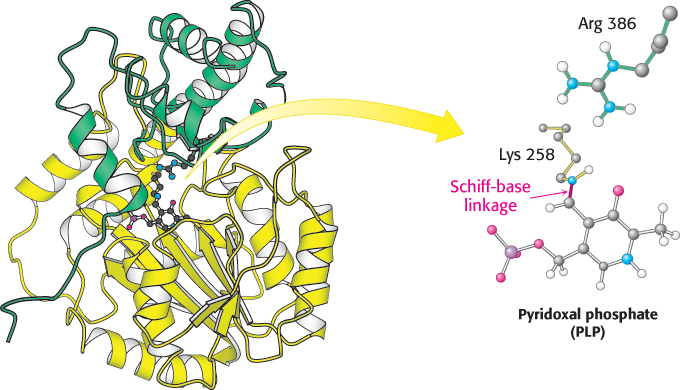
691
Blood levels of aminotransferases serve a diagnostic function
 The presence of alanine and aspartate aminotransferase in the blood is an indication of liver damage. Liver damage can occur for a number of reasons, including viral hepatitis, long-
The presence of alanine and aspartate aminotransferase in the blood is an indication of liver damage. Liver damage can occur for a number of reasons, including viral hepatitis, long-
Pyridoxal phosphate enzymes catalyze a wide array of reactions
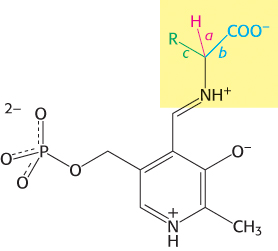
Transamination is just one of a wide range of amino acid transformations that are catalyzed by PLP enzymes. The other reactions catalyzed by PLP enzymes at the α-carbon atom of amino acids are decarboxylations, deaminations, racemizations, and aldol cleavages (Figure 23.11). In addition, PLP enzymes catalyze elimination and replacement reactions at the β-carbon atom (e.g., tryptophan synthetase in the synthesis of tryptophan) and the γ-carbon atom (e.g., cystathionine β-synthase in the synthesis of cysteine) of amino acid substrates. Three common features of PLP catalysis underlie these diverse reactions.
A Schiff base is formed by the amino acid substrate (the amine component) and PLP (the carbonyl component).
The protonated form of PLP acts as an electron sink to stabilize catalytic intermediates that are negatively charged. Electrons from these intermediates are attracted to the positive charge on the ring nitrogen atom. In other words, PLP is an electrophilic catalyst.
The product Schiff base is cleaved at the completion of the reaction.
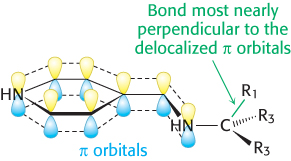
How does an enzyme selectively break a particular one of three bonds at the α-carbon atom of an amino acid substrate? An important principle is that the bond being broken must be perpendicular to the π orbitals of the electron sink (Figure 23.12). An aminotransferase, for example, binds the amino acid substrate so that the Cα—H bond is perpendicular to the PLP ring (Figure 23.13). In serine hydroxymethyltransferase, the enzyme that converts serine into glycine, the N—
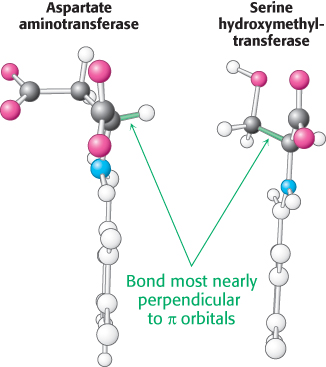
692
 Many of the PLP enzymes that catalyze amino acid transformations, such as serine hydroxymethyltransferase, have a similar structure and are clearly related by divergent evolution. Others, such as tryptophan synthetase, have quite different overall structures. Nonetheless, the active sites of these enzymes are remarkably similar to that of aspartate aminotransferase, revealing the effects of convergent evolution.
Many of the PLP enzymes that catalyze amino acid transformations, such as serine hydroxymethyltransferase, have a similar structure and are clearly related by divergent evolution. Others, such as tryptophan synthetase, have quite different overall structures. Nonetheless, the active sites of these enzymes are remarkably similar to that of aspartate aminotransferase, revealing the effects of convergent evolution.
Serine and threonine can be directly deaminated
The α-amino groups of serine and threonine can be directly converted into NH4+ without first being transferred to α-ketoglutarate. These direct deaminations are catalyzed by serine dehydratase and threonine dehydratase, in which PLP is the prosthetic group.
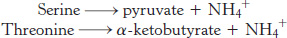
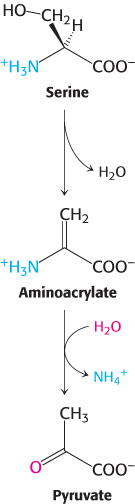
These enzymes are called dehydratases because dehydration precedes deamination. Serine loses a hydrogen ion from its α-carbon atom and a hydroxide ion group from its β-carbon atom to yield aminoacrylate. This unstable compound reacts with H2O to give pyruvate and NH4+. Thus, the presence of a hydroxyl group attached to the β-carbon atom in each of these amino acids permits the direct deamination.
Peripheral tissues transport nitrogen to the liver
Although most amino acid degradation takes place in the liver, other tissues can degrade amino acids. For instance, muscle uses branched-
Nitrogen is transported from muscle to the liver in two principal transport forms. Glutamate is formed by transamination reactions, but the nitrogen is then transferred to pyruvate to form alanine, which is released into the blood (Figure 23.14). The liver takes up the alanine and converts it back into pyruvate by transamination. The pyruvate can be used for gluconeogenesis and the amino group eventually appears as urea. This transport is referred to as the glucose–
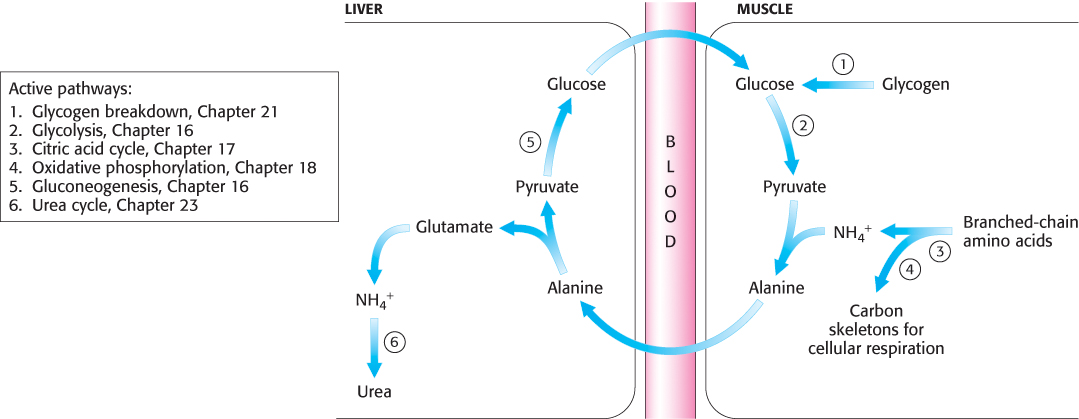
693
Glutamine is also a key transport form of nitrogen. Glutamine synthetase catalyzes the synthesis of glutamine from glutamate and NH4+ in an ATP-
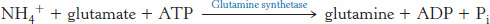
The nitrogens of glutamine can be converted into urea in the liver.