26.1 Phosphatidate Is a Common Intermediate in the Synthesis of Phospholipids and Triacylglycerols
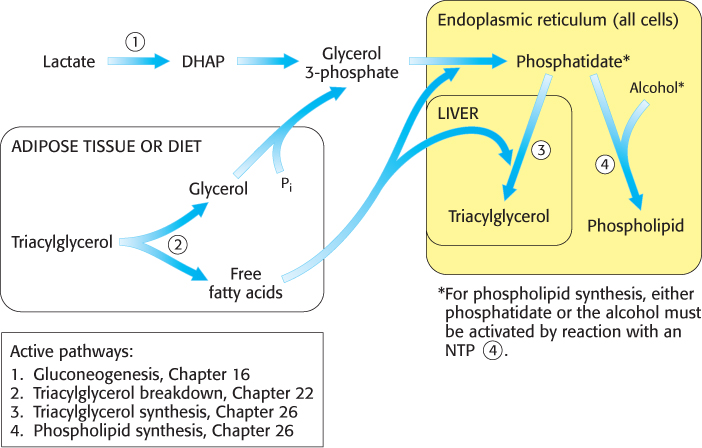
Lipid synthesis requires the coordinated action of gluconeogenesis and fatty acid metabolism, as illustrated in Figure 26.1. The common step in the synthesis of both phospholipids for membranes and triacylglycerols for energy storage is the formation of phosphatidate (diacylglycerol 3-
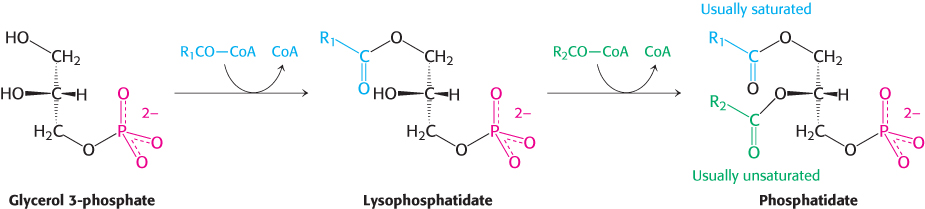
These acylations are catalyzed by glycerol phosphate acyltransferase. In most phosphatidates, the fatty acid chain attached to the C-
Diacylglycerol + ATP → phosphatidate + ADP
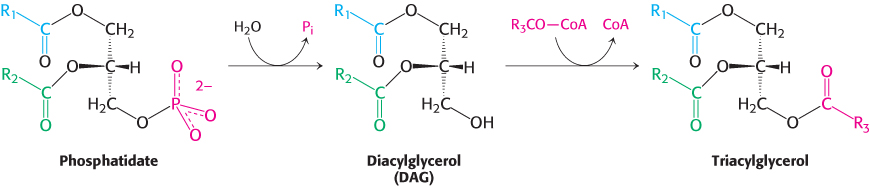
The phospholipid and triacylglycerol pathways diverge at phosphatidate. In the synthesis of triacylglycerols, a key enzyme in the regulation of lipid synthesis, phosphatidic acid phosphatase, hydrolyzes phosphatidate to give a diacylglycerol. This intermediate is acylated to a triacylglycerol through the addition of a third fatty acid chain in a reaction that is catalyzed by diglyceride acyltransferase. Both enzymes are associated in a triacylglycerol synthetase complex that is bound to the endoplasmic reticulum membrane.
769
The liver is the primary site of triacylglycerol synthesis. From the liver, the triacylglycerols are transported to the muscles for energy conversion or to the adipose cells for storage.
The synthesis of phospholipids requires an activated intermediate
Membrane-
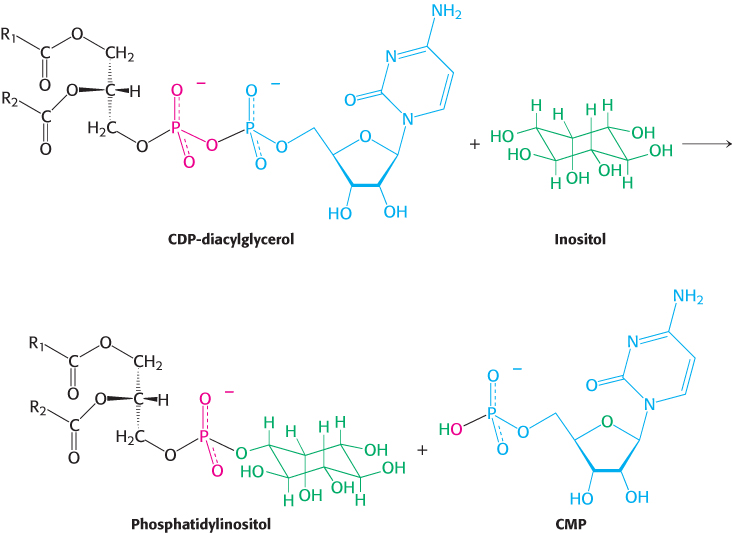
The synthesis of some phospholipids begins with the reaction of phosphatidate with cytidine triphosphate (CTP) to form the activated diacylglycerol, cytidine diphosphodiacylglycerol (CDP-
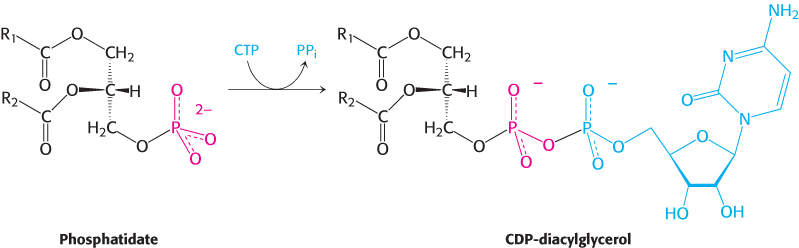
The activated phosphatidyl unit then reacts with the hydroxyl group of an alcohol to form a phosphodiester linkage. If the alcohol is inositol, the products are phosphatidylinositol and cytidine monophosphate (CMP).
770
Subsequent phosphorylations catalyzed by specific kinases lead to the synthesis of phosphatidylinositol 4,5-
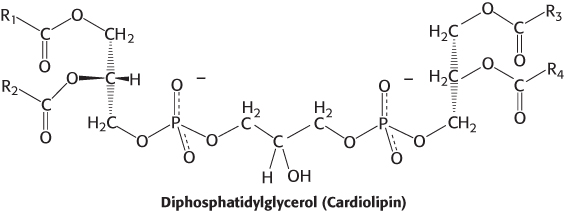
The fatty acid components of phospholipids may vary, and thus cardiolipin, as well as most other phospholipids, represents a class of molecules rather than a single species. As a result, a single mammalian cell may contain thousands of distinct phospholipids. Phosphatidylinositol is unusual in that it has a nearly fixed fatty acid composition. Stearic acid usually occupies the C-
Some phospholipids are synthesized from an activated alcohol
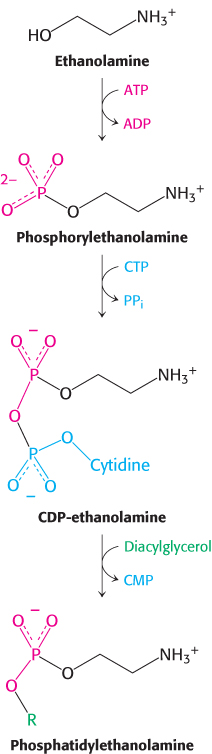
Phosphatidylethanolamine, the major phospholipid of the inner leaflet of cell membranes, is synthesized from the alcohol ethanolamine. To activate the alcohol, ethanolamine is phosphorylated by ATP to form the precursor, phosphorylethanolamine. This precursor then reacts with CTP to form the activated alcohol, CDP-
Phosphatidylcholine is an abundant phospholipid
The most common phospholipid in mammals is phosphatidylcholine, comprising approximately 50% of the membrane mass. Dietary choline is activated in a series of reactions analogous to those in the activation of ethanolamine. CTP-
 Earlier (Section 22.4) we examined how cancer cells increase lipogenesis to meet the fatty acid needs for membrane synthesis. Evidence is accumulating that CCT is specifically activated in some cancers to generate the required phosphocholine.
Earlier (Section 22.4) we examined how cancer cells increase lipogenesis to meet the fatty acid needs for membrane synthesis. Evidence is accumulating that CCT is specifically activated in some cancers to generate the required phosphocholine.
771
The importance of phosphatidylcholine is attested to by the fact that the liver possesses an enzyme, phosphatidylethanolamine methyltransferase, which synthesizes phosphatidylcholine from phosphatidylethanolamine when dietary choline is insufficient. The amino group of this phosphatidylethanolamine is methylated three times to form phosphatidylcholine. S-
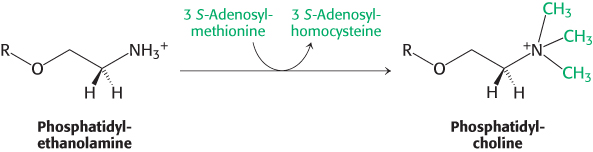
Thus, phosphatidylcholine can be produced by two distinct pathways in mammals, ensuring that this phospholipid can be synthesized even if the components for one pathway are in limited supply.
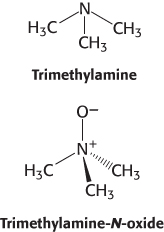
Excess choline is implicated in the development of heart disease
Choline is a popular dietary supplement that is believed by some to enhance liver and neuronal function. Although the effectiveness of choline supplements is not established, the dangers of excess choline consumption are becoming clear. Gut bacteria convert excess choline into trimethylamine (TMA), a gas that smells like rotten fish, and the liver converts the absorbed TMA into trimethylamine-
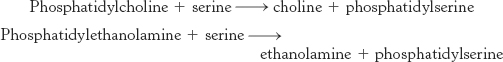
Base-exchange reactions can generate phospholipids
Phosphatidylserine makes up 10% of the phospholipids in mammals. This phospholipid is synthesized in a base-
Phosphatidylserine is normally located in the inner leaflet of the plasma membrane bilayer but is moved to the outer leaflet in apoptosis (Section 18.6). There, it serves to attract phagocytes to consume the cell remnants after apoptosis is complete. Phosphatidylserine is translocated from one side of the membrane to the other by an ATP-
Note that a cytidine nucleotide plays the same role in the synthesis of these phosphoglycerides as a uridine nucleotide does in the formation of glycogen (Section 21.4). In all of these biosyntheses, an activated intermediate (UDP-
772
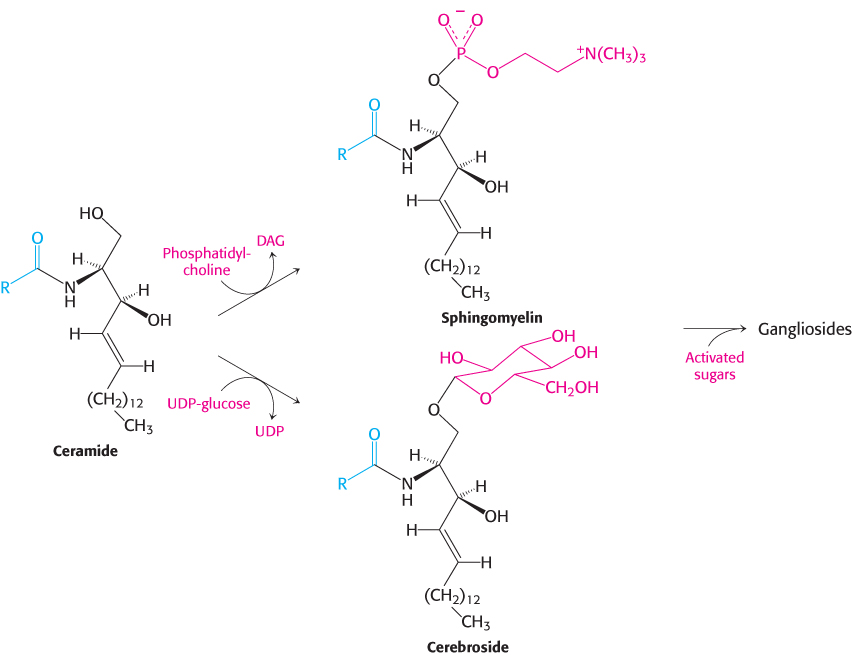
Sphingolipids are synthesized from ceramide
We now turn from glycerol-
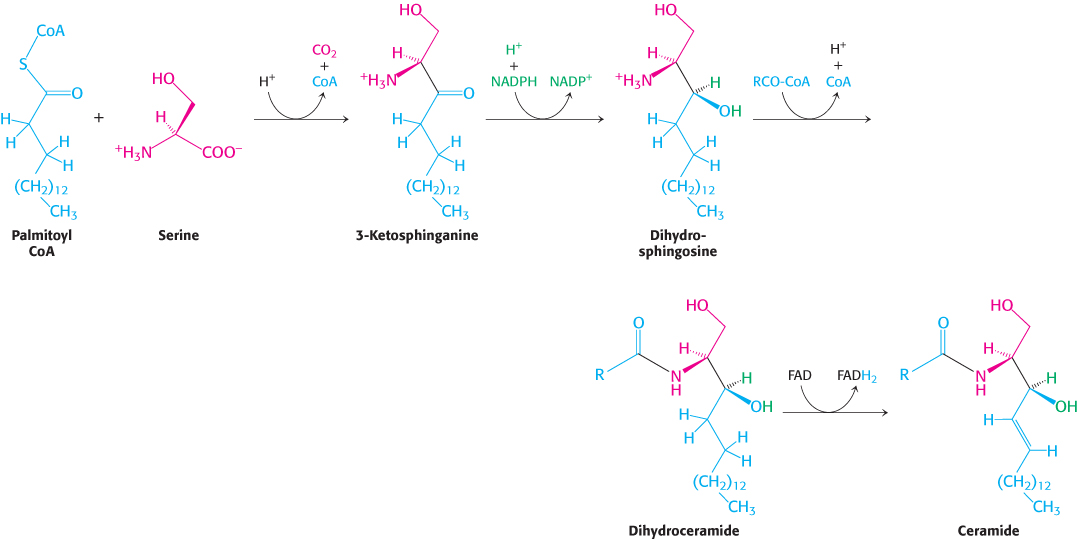
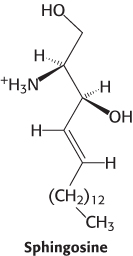
In all sphingolipids, the amino group of ceramide is acylated. The terminal hydroxyl group also is substituted (Figure 26.3). In sphingomyelin, a component of the myelin sheath covering many nerve fibers, the substituent is phosphorylcholine, which comes from phosphatidylcholine. In a cerebroside, the substituent is glucose or galactose. UDP-
Gangliosides are carbohydrate-rich sphingolipids that contain acidic sugars
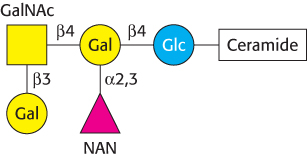
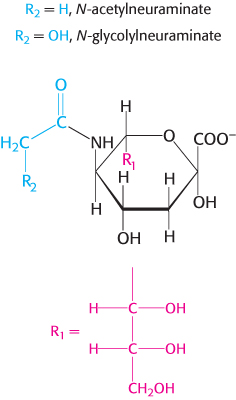
Gangliosides are the most complex sphingolipids. In a ganglioside, an oligosaccharide chain is linked to the terminal hydroxyl group of ceramide by a glucose residue (Figure 26.4). This oligosaccharide chain contains at least one acidic sugar, either N-
Gangliosides are synthesized by the ordered, step-
773
Ganglioside-
Sphingolipids confer diversity on lipid structure and function
The structures of sphingolipids and the more abundant glycerophospholipids are very similar (Figure 12.8). Given the structural similarity of these two types of lipids, why are sphingolipids required at all? Indeed, the prefix “sphingo” was applied to capture the “sphinxlike” properties of this enigmatic class of lipids. Although the precise role of sphingolipids is not firmly established, progress toward solving the riddle of their function is being made. As discussed in Chapter 12, sphingolipids are important components of lipid rafts, highly organized regions of the plasma membrane that are important in signal transduction. Sphingosine, sphingosine 1-
774
Respiratory distress syndrome and Tay–Sachs disease result from the disruption of lipid metabolism
Respiratory distress syndrome is a pathological condition resulting from a failure in the biosynthetic pathway for dipalmitoylphosphatidylcholine. This phospholipid, in conjunction with specific proteins and other phospholipids, is found in the extracellular fluid that surrounds the alveoli of the lung. Its function is to decrease the surface tension of the fluid to prevent lung collapse at the end of the expiration phase of breathing. Premature infants may suffer from respiratory distress syndrome because their immature lungs do not synthesize enough dipalmitoylphosphatidylcholine.
Tay–Sachs disease is caused by a failure of lipid degradation: an inability to degrade gangliosides. Gangliosides are found in highest concentration in the nervous system, particularly in gray matter, where they constitute 6% of the lipids. Gangliosides are normally degraded inside lysosomes by the sequential removal of their terminal sugars but, in Tay–
The ganglioside content of the brain of an infant with Tay–
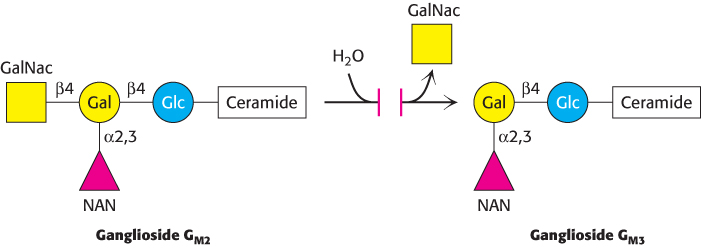
Tay–
Ceramide metabolism stimulates tumor growth
Ceramide is a precursor for sphingomyelin, cerebroside, and gangliosides. Ceramide itself, however, induces programmed cell death or apoptosis (Section 18.6). Recall that cancer cells require all types of lipids for membrane formation (Section 22.4). How do cancer cells prevent ceramide-
775
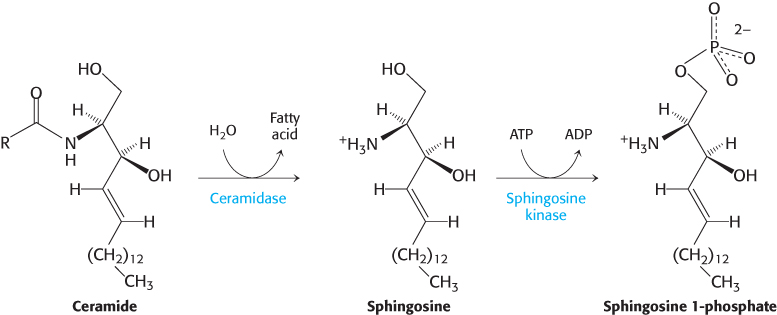
Thus, cancer cells convert a potentially lethal signal molecule into one that promotes tumor growth. Efforts are underway to develop inhibitors of ceramidase for use as chemotherapeutic agents.
Phosphatidic acid phosphatase is a key regulatory enzyme in lipid metabolism
Although the details of the regulation of lipid synthesis remain to be elucidated, evidence suggests that phosphatidic acid phosphatase (PAP), working in concert with diacylglycerol kinase, plays a key role in the regulation of lipid synthesis. PAP, also called lipin 1 in mammals, controls the extent to which triacylglycerols are synthesized relative to phospholipids and regulates the type of phospholipid synthesized (Figure 26.6). For instance, when PAP activity is high, phosphatidate is dephosphorylated and diacylglycerol is produced, which can react with the appropriate activated alcohols to yield phosphatidylethanolamine, phosphatidylserine, or phosphatidylcholine. Diacylglycerol can also be converted into triacylglycerols, and evidence suggests that the formation of triacylglycerols may act as a fatty acid buffer. This buffering helps to regulate the levels of diacylglycerol and sphingolipids, which serve signaling functions.
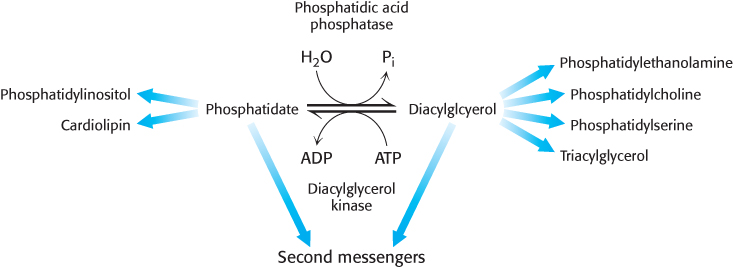
When PAP activity is lower, phosphatidate is used as a precursor for different phospholipids, such as phosphatidylinositol and cardiolipin. Moreover, phosphatidate is a signal molecule itself. Phosphatidate regulates the growth of endoplasmic reticulum and nuclear membranes and acts as a cofactor that stimulates the expression of genes in phospholipid synthesis.
776
What are the signal molecules that regulate the activity of PAP? CDP-
Studies in mice clearly show the importance of PAP for the regulation of fatty acid synthesis. The loss of PAP function prevents normal adipose-