27.3 !dna! Diabetes Is a Common Metabolic Disease Often Resulting from Obesity
Diabetes
Named for the excessive urination in the disease. Aretaeus, a Cappadocian physician of the second century A.D., wrote: “The epithet diabetes has been assigned to the disorder, being something like passing of water by a siphon.” He perceptively characterized diabetes as “being a melting-down of the flesh and limbs into urine.”
From Latin, meaning “sweetened with honey.” Refers to the presence of sugar in the urine of patients having the disease. Mellitus distinguishes this disease from diabetes insipidus, which is caused by impaired renal reabsorption of water.
Having taken an overview of the regulation of body weight, we now examine the biochemical results when regulation fails because of behavior, genetics, or a combination of the two. The most common result of such a failure is obesity, a condition in which excess energy is stored as triacylglycerols. Recall that all excess food consumption is ultimately converted into triacylglycerols. Humans maintain about a day’s worth of glycogen and, after these stores have been replenished, excess carbohydrates are converted into fats and then into triacylglycerols. Amino acids are not stored at all, and so excess amino acids are ultimately converted into fats also. Thus, regardless of the type of food consumed, excess consumption results in increased fat stores.
We begin our consideration of the effects of disruptions in caloric homeostasis with diabetes mellitus, a complex disease characterized by grossly abnormal fuel usage: glucose is overproduced by the liver and underutilized by other organs. The incidence of diabetes mellitus (usually referred to simply as diabetes) is about 5% of the world population. Type 1 diabetes is caused by the autoimmune destruction of the insulin-secreting β cells in the pancreas and usually begins before age 20. Type 1 diabetes is also referred to as insulin-dependent diabetes, meaning that the affected person requires the administration of insulin to live. Most diabetics, in contrast, have a normal or even higher level of insulin in their blood, but they are unresponsive to the hormone, a characteristic called insulin resistance. This form of the disease, known as type 2 diabetes, typically arises later in life than does the insulin-dependent form. Type 2 diabetes accounts for approximately 90% of the diabetes cases throughout the world and is the most common metabolic disease in the world. In the United States, it is the leading cause of blindness, kidney failure, and amputation. Obesity is a significant predisposing factor for the development of type 2 diabetes.
Insulin initiates a complex signal-transduction pathway in muscle
What is the biochemical basis of insulin resistance? How does insulin resistance lead to failure of the β cells of the pancreas that results in type 2 diabetes? How does obesity contribute to this progression? To answer these questions and begin to unravel the mysteries of metabolic disorders, let us examine the mechanism of action of insulin in muscle, the largest tissue target of insulin. Indeed, muscle uses about 85% of the glucose ingested during a meal.
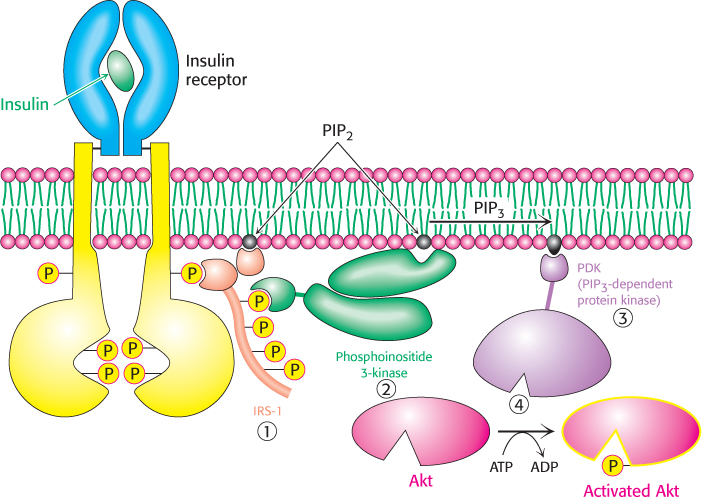
Figure 27.5: Insulin signaling. The binding of insulin results in the cross-phosphorylation and activation of the insulin receptor. Phosphorylated sites on the receptor act as binding sites for insulin-receptor substrates such as IRS-1. The lipid kinase phosphoinositide 3-kinase binds to phosphorylated sites on IRS-1 through its regulatory domain and then converts PIP2 into PIP3. Binding to PIP3 activates PIP3-dependent protein kinase, which phosphorylates and activates kinases such as Akt. Activated Akt can then diffuse throughout the cell and continue the signal-transduction pathway.
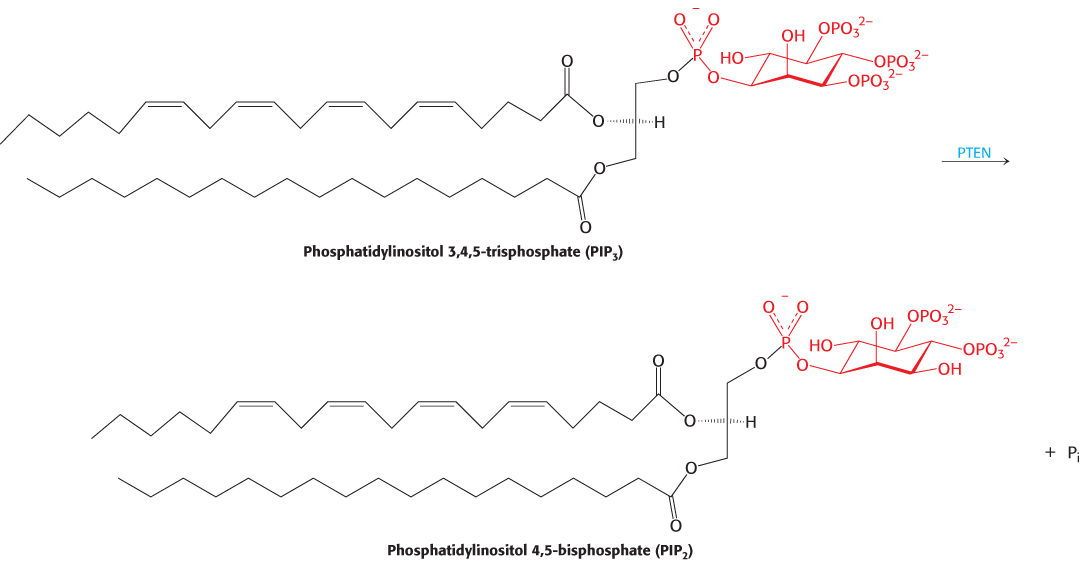
In a normal cell, insulin binds to its receptor, which then autophosphorylates on tyrosine residues, with each subunit of the receptor phosphorylating its partner. Phosphorylation of the receptor generates binding sites for insulin-receptor substrates (IRSs), such as IRS-1 (Figure 27.5). Subsequent phosphorylation of IRS-1 by the tyrosine kinase activity of the insulin receptor engages the insulin-signaling pathway (1). Phosphorylated IRS-1 binds to phosphoinositide 3-kinase (PI3K) and activates it. PI3K catalyzes the conversion of phosphatidylinositol 4,5-bisphosphate (PIP2) into phosphatidylinositol 3,4,5-trisphosphate (PIP3), a second messenger (2). PIP3 activates the phosphatidylinositol-dependent protein kinase (PDK) (3), which in turn activates several other kinases, most notably Akt (4), also known as protein kinase B (PKB). Akt facilitates the translocation of the glucose transporter (GLUT4)-containing vesicles to the cell membrane, which leads to a more robust absorption of glucose from the blood. Moreover, Akt phosphorylates and inhibits glycogen synthase kinase (GSK3). Recall that GSK3 inhibits glycogen synthase (Section 21.5). Thus, insulin leads to the absorption of glucose from the blood, activation of glycogen synthase, and enhanced glycogen synthesis.
Like all signal pathways, the insulin-signaling cascade must be capable of being turned off. Three different processes contribute to the down-regulation of insulin signaling. First, phosphatases deactivate the insulin receptor and destroy a key second messenger. Tyrosine phosphatase IB removes phosphoryl groups from the receptor, thus inactivating it. The second messenger PIP3 is inactivated by the phosphatase PTEN (phosphatase and tensin homolog), which dephosphorylates it, forming PIP2, which itself has no second-messenger properties.
Second, the IRS protein can be inactivated by phosphorylation on serine residues by specific Ser/Thr kinases. These kinases are activated by overnutrition and other stress signals and may play a role in the development of insulin resistance. Finally, SOCS proteins, the regulatory proteins discussed earlier, interact with the insulin receptor and IRS-1 and apparently facilitate their proteolytic degradation by the proteasome complex.
Metabolic syndrome often precedes type 2 diabetes
With our knowledge of the key components of energy homeostasis, let us begin our investigation of the biochemical basis of insulin resistance and type 2 diabetes. Obesity is a contributing factor to the development of insulin resistance, which is an early development on the path to type 2 diabetes. Indeed, a cluster of pathologies—including insulin resistance, hyperglycemia, dyslipidemia (high blood levels of triacylglycerols, cholesterol, and low-density lipoproteins)—often develop together. This clustering, called metabolic syndrome, is thought to be a predecessor of type 2 diabetes.
A consequence of obesity is that the amount of triacylglycerols consumed exceeds the adipose tissue’s storage capacity. As a result, other tissues begin to accumulate fat, most notably liver and muscle (Figure 27.6). For reasons to be presented later in the chapter, this accumulation results in insulin resistance and ultimately in pancreatic failure. We will focus on muscle and the β cells of the pancreas.
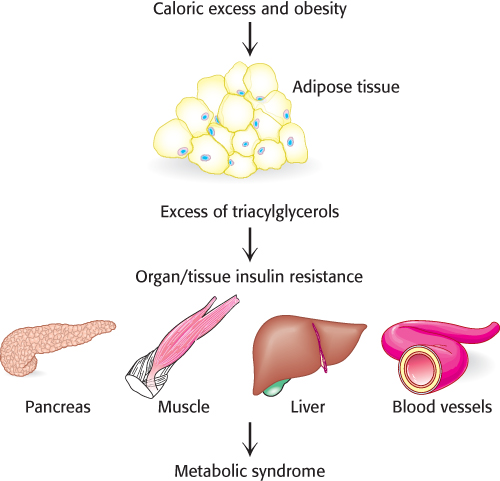
Figure 27.6: The storage capacity of fat tissue can be exceeded in obesity. In caloric excess, the storage capacity of adipocytes can be exceeded with deleterious results. The excess fat accumulates in other tissues, resulting in biochemical malfunction of the tissues. When the pancreas, muscle, liver, and cells lining the blood vessels are affected, metabolic syndrome, a condition that often precedes type 2 diabetes, may result.
[Information from S. Fröjdö, H. Vidal, and L. Pirola, Biochim. Biophys. Acta 1792:83–92, 2009, Fig. 1.]
Excess fatty acids in muscle modify metabolism
We have seen many times the importance of fats as fuels. In regard to obesity, more fats are present than can be processed by muscle. Although the rate of β oxidation increases in response to the high concentration of fats, mitochondria are not capable of processing all of the fatty acids by β oxidation; fatty acids accumulate in the mitochondria and eventually spill over into the cytoplasm. Indeed, the inability to process all of the fatty acids results in their reincorporation into triacylglycerols and the accumulation of fat in the cytoplasm. In the cytoplasm, levels of diacylglycerol and ceramide (a component of sphingolipids) also increase. Diacylglycerol is a second messenger that activates protein kinase C (PKC) (Section 14.1). When active, PKC and other Ser/Thr protein kinases are capable of phosphorylating IRS and reducing the ability of IRS to propagate the insulin signal. Saturated and trans unsaturated fatty acids may also activate kinases that block the insulin signal. Ceramide or its metabolites inhibit glucose uptake and glycogen synthesis, apparently by inhibiting PDK and Akt. The result is a diet-induced insulin resistance (Figure 27.7).
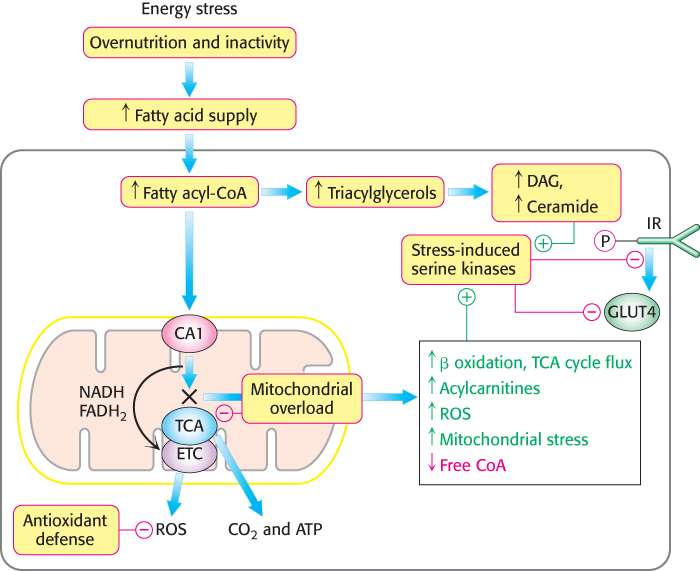
Figure 27.7: Excess fat in peripheral tissues can result in insulin insensitivity. Excess fat accumulation in peripheral tissues, most notably muscle, can disrupt some signal-transduction pathways and inappropriately activate others. In particular, diacylglycerols and ceramide activate stress-induced pathways that interfere with insulin signaling, resulting in insulin resistance. (Abbreviations: DAG, diacylglycerol; TGs, triacylglycerols; ROS, reactive oxygen species; CA1, carnitine acyltransferase 1; GLUT4, glucose transporter 4; IR, insulin receptor; ETC, electron-transport chain; TCA, tricarboxylic acid cycle.)
Insulin resistance in muscle facilitates pancreatic failure
What is the effect of overnutrition on the pancreas? This question is important because a primary function of the pancreas is to respond to the presence of glucose in the blood by secreting insulin, a process referred to as glucose-stimulated insulin secretion (GSIS). Indeed, the β cell is a virtual insulin factory. Proinsulin mRNA, which encodes proinsulin, the precursor to insulin, constitutes 20% of the total mRNA in the pancreas. Proinsulin constitutes 50% of the total protein synthesized in the pancreas.
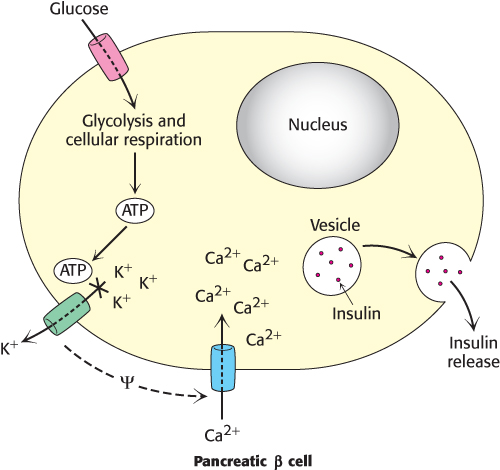
Figure 27.8: Insulin release is regulated by ATP. The metabolism of glucose by glycolysis and cellular respiration increases the concentration of ATP, which causes an ATP-sensitive potassium channel to close. The closure of this channel alters the charge across the membrane (ψ) and causes a calcium channel to open. The influx of calcium causes insulin-containing granules to fuse with the plasma membrane, releasing insulin into the blood.
Glucose enters the β cells of the pancreas through the glucose transporter GLUT2. Recall that GLUT2 will allow glucose transport only when blood glucose is plentiful, ensuring that insulin is secreted only when glucose is abundant, such as after a meal (Section 16.2). The β cell metabolizes glucose to CO2 and H2O in the process of cellular respiration, generating ATP (Chapters 16, 17, and 18). The resulting increase in the ATP/ADP ratio closes an ATP-sensitive K+ channel that, when open, allows potassium to flow out of the cell (Figure 27.8). The resulting alteration in the cellular ionic environment opens a Ca2+ channel. The influx of Ca2+ causes insulin-containing secretory vesicles to fuse with the cell membrane and release insulin into the blood. Thus, the increase in energy charge resulting from the metabolism of glucose has been translated by the membrane proteins into a physiological response—the secretion of insulin and the subsequent removal of glucose from the blood.
What aspect of β cell function ultimately fails as a result of overnutrition, causing the transition from insulin resistance to full-fledged type 2 diabetes? Recall that, under normal circumstances, the β cells of the pancreas synthesize large amounts of proinsulin. The proinsulin folds into its three-dimensional structure in the endoplasmic reticulum, is processed to insulin, and is subsequently packaged into vesicles for secretion. As insulin resistance develops in the muscle, the β cells respond by synthesizing yet more insulin in a futile attempt to drive insulin action. The ability of the endoplasmic reticulum to process all of the proinsulin and insulin becomes compromised, a condition known as endoplasmic reticulum (ER) stress, and unfolded or misfolded proteins accumulate. ER stress initiates a signal pathway called the unfolded protein response (UPR), a pathway intended to save the cell. UPR consists of several steps. First, general protein synthesis is inhibited so as to prevent more proteins from entering the ER. Second, chaperone synthesis is stimulated. Recall that chaperones are proteins that assist the folding of other proteins (Section 2.6). Third, misfolded proteins are removed from the ER and are subsequently delivered to the proteasome for destruction. Finally, if the described response fails to alleviate the ER stress, programmed cell death is triggered, which ultimately leads to β cell death and full-fledged type 2 diabetes.
What are the treatments for type 2 diabetes? Most are behavioral in nature. Diabetics are advised to count calories, making sure that energy intake does not exceed energy output; to consume a diet rich in vegetables, fruits, and grains; and to get plenty of aerobic exercise. Note that these guidelines are the same as those for healthy living, even for those not suffering from type 2 diabetes. Treatments specific for type 2 diabetes include the monitoring of blood-glucose concentration so that it is within the target range (normal is 3.6 to 6.1 mM). For those who are not able to maintain proper glucose levels with the behaviors described herein, drug treatments are required. The administration of insulin may be necessary upon pancreatic failure, and treatment with metformin (Glucophage), which activates AMPK, may be effective. AMPK promotes the oxidation of fats while inhibiting fat synthesis and storage. It also stimulates glucose uptake and storage by muscle while inhibiting gluconeogenesis in the liver.
Metabolic derangements in type 1 diabetes result from insulin insufficiency and glucagon excess
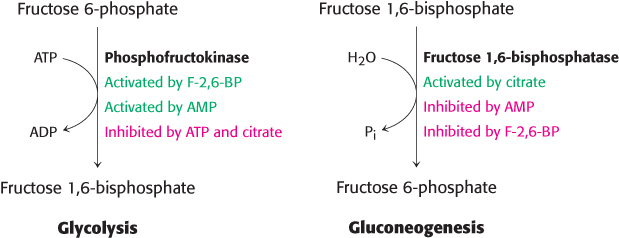
Figure 27.9: Regulation of glycolysis and gluconeogenesis. Phosphofructokinase is the key enzyme in the regulation of glycolysis, whereas fructose 1,6-bisphosphatase is the principal enzyme controlling the rate of gluconeogenesis. Note the reciprocal relation between the pathways and the signal molecules.
We now turn to the more-straightforward type 1 diabetes. In type 1 diabetes, insulin production is insufficient because of autoimmune destruction of the β cells of the pancreas. Consequently, the glucagon/insulin ratio is at higher-than-normal levels. In essence, the diabetic person is in biochemical fasting mode despite a high concentration of blood glucose. Because insulin is deficient, the entry of glucose into adipose and muscle cells is impaired. The liver becomes stuck in a gluconeogenic and ketogenic state. The gluconeogenic state is characterized by excessive production of glucose. The excessive level of glucagon relative to that of insulin leads to a decrease in the amount of fructose 2,6-bisphosphate (F-2, 6-BP), which stimulates glycolysis and inhibits gluconeogenesis in the liver. Hence, glycolysis is inhibited and gluconeogenesis is stimulated because of the opposite effects of F-2, 6-BP on phosphofructokinase and fructose-1, 6-bisphosphatase (Section 16.4; Figure 27.9). Essentially, the cells’ response to a lack of insulin amplifies the amount of glucose in the blood. The high glucagon/insulin ratio in diabetes also promotes glycogen breakdown. Hence, an excessive amount of glucose is produced by the liver and released into the blood. Glucose is excreted in the urine (hence the name mellitus) when its concentration in the blood exceeds the reabsorptive capacity of the renal tubules. Water accompanies the excreted glucose, and so an untreated diabetic in the acute phase of the disease is hungry and thirsty.
Because carbohydrate utilization is impaired, a lack of insulin leads to the uncontrolled breakdown of lipids and proteins, resulting in the ketogenic state. Large amounts of acetyl CoA are then produced by β oxidation. However, much of the acetyl CoA cannot enter the citric acid cycle, because there is insufficient oxaloacetate for the condensation step. Recall that mammals can synthesize oxaloacetate from pyruvate, a product of glycolysis, but not from acetyl CoA; instead, they generate ketone bodies (Section 22.3). A striking feature of diabetes is the shift in fuel usage from carbohydrates to fats; glucose, more abundant than ever, is spurned. In high concentrations, ketone bodies overwhelm the kidney’s capacity to maintain acid–base balance. The untreated diabetic can go into a coma because of a lowered blood-pH level and dehydration. Interestingly, diabetic ketosis is rarely a problem in type 2 diabetes because insulin is active enough to prevent excessive lipolysis in liver and adipose tissue.
What is the treatment for type 1 diabetes? Many of the behaviors recommended for type 2 diabetes apply to type 1: watching calories, exercising, and eating a healthy diet. Likewise, blood-glucose levels must be monitored. Insulin treatments are required for survival.