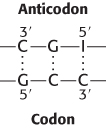
30.1 Protein Synthesis Requires the Translation of Nucleotide Sequences into Amino Acid Sequences
The basics of protein synthesis are the same across all kingdoms of life—evidence that the protein-synthesis system arose very early in evolution. An mRNA is decoded, or read, in the 5′-to-3′ direction, one codon at a time, and the corresponding protein is synthesized in the amino-to-carboxyl direction by the sequential addition of amino acids to the carboxyl end of the growing peptide chain (Figure 30.1). The amino acids arrive at the growing chain in activated form as aminoacyl-tRNAs, created by joining the carboxyl group of an amino acid to the 3′ end of a tRNA molecule. The linking of an amino acid to its corresponding tRNA is catalyzed by an aminoacyl-tRNA synthetase. For each amino acid, there is usually one activating enzyme and at least one kind of tRNA.
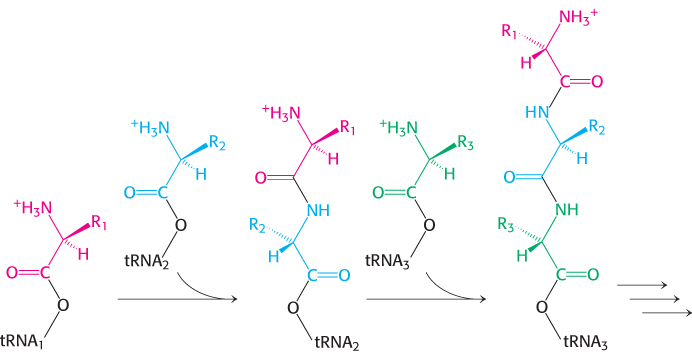
Figure 30.1: Polypeptide-chain growth. Proteins are synthesized by the successive addition of amino acids to the carboxyl terminus.
The synthesis of long proteins requires a low error frequency
The process of transcription is analogous to copying, word for word, a page of a book. There is no change of alphabet or vocabulary; so the likelihood of a change in meaning is small. Translating the base sequence of an mRNA molecule into a sequence of amino acids is analogous to translating the page of a book into another language. Translation is a complex process, entailing many steps and dozens of molecules. The potential for error exists at each step. The complexity of translation creates a conflict between two requirements: the process must be both accurate and fast enough to meet a cell’s needs. In E. coli, translation can take place at a rate of 20 amino acids per second, a truly impressive speed considering the complexity of the process.
How accurate must protein synthesis be? Let us consider error rates. The probability of forming a protein with no errors depends on the number of amino acid residues and on the frequency (ε) of insertion of a wrong amino acid. As Table 30.1 shows, an error frequency of 10−2 is intolerable, even for quite small proteins. An ε value of 10−3 usually leads to the error-free synthesis of a 300-residue protein (~33 kDa) but not of a 1000-residue protein (~110 kDa). Thus, the error frequency must not exceed approximately 10−4 to produce the larger proteins effectively. Lower error frequencies are conceivable; however, except for the largest proteins, they will not dramatically increase the percentage of proteins with accurate sequences. In addition, such lower error rates are likely to be possible only by a reduction in the rate of protein synthesis because additional time for proofreading is required. In fact, the observed values of ε are close to 10−4. An error frequency of about 10−4 per amino acid residue was selected in the course of evolution to accurately produce proteins consisting of as many as 1000 amino acids while maintaining a remarkably rapid rate for protein synthesis.
|
|
Probability of synthesizing an error-free protein |
|
|
Number of amino acid residues |
Frequency of inserting an incorrect amino acid |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Note: The probability p of forming a protein with no errors depends on n, the number of amino acids, and ε, the frequency of insertion of a wrong amino acid: p = (1 − ε)n. |
Table 30.1: Accuracy of protein synthesis
Transfer RNA molecules have a common design
The fidelity of protein synthesis requires accurate recognition of three-base codons on messenger RNA. Recall that the genetic code relates each amino acid to a three-letter codon (Section 4.6). An amino acid cannot itself recognize a codon. Consequently, an amino acid is attached to a specific tRNA molecule that recognizes the codon by Watson–Crick base-pairing. Transfer RNA serves as the adapter molecule that binds to a specific codon and brings with it an amino acid for incorporation into the polypeptide chain.
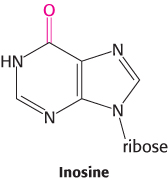
Consider yeast alanyl-tRNA, so called because it will carry the amino acid alanine. This adapter molecule is a single chain of 76 ribonucleotides (Figure 30.2). The 5′ terminus is phosphorylated (pG), whereas the 3′ terminus has a free hydroxyl group. The amino acid-attachment site is the 3′-hydroxyl group of the adenosine residue at the 3′ terminus of the molecule. The sequence 5′-IGC-3′ in the middle of the molecule is the anticodon, where I is the purine base inosine. It is complementary to 5′-GCC-3′, one of the codons for alanine.
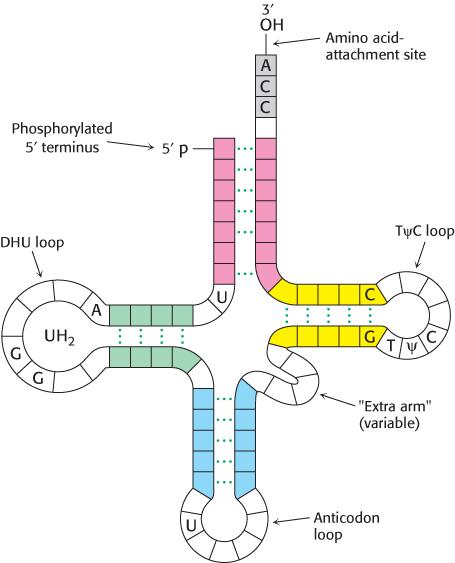
Figure 30.3: General structure of tRNA molecules. Comparison of the base sequences of many tRNAs reveals a number of conserved features.
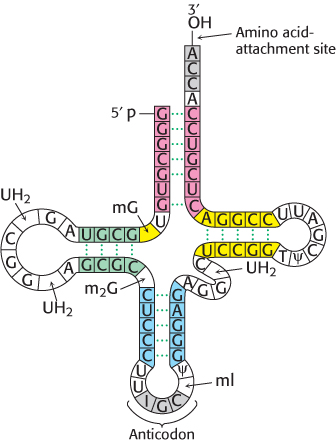
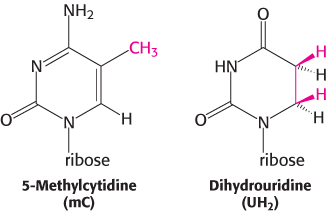
Figure 30.2: Alanyl-tRNA sequence. The base sequence of yeast alanyl-tRNA and the deduced cloverleaf secondary structure are shown. Modified nucleosides are abbreviated as follows: methylinosine (ml), dihydrouridine (UH2), ribothymidine (T), pseudouridine (ψ), methylguanosine (mG), and (dimethylguanosine (m2G). Inosine (l), another modified nucleoside, is part of the anticodon.
Thousands of tRNA sequences are known. The striking finding is that all of them can be arranged in a cloverleaf pattern in which about half the residues are base-paired (Figure 30.3). Hence, tRNA molecules have many common structural features. This finding is not unexpected, because all tRNA molecules must be able to interact in nearly the same way with the ribosomes, mRNAs, and protein factors that participate in translation.
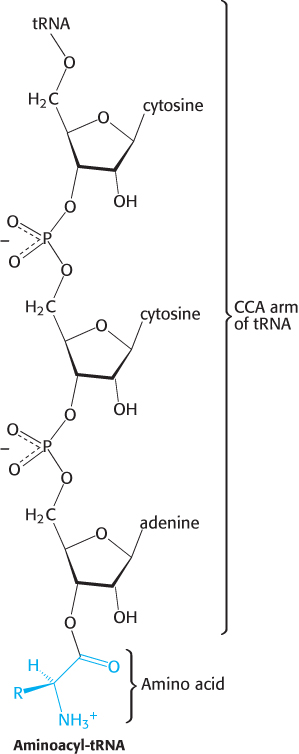
Figure 30.6: Aminoacyl-tRNA. Amino acids are coupled to tRNAs through ester linkages to either the 2′- or the 3′-hydroxyl group of the 3′-adenosine residue. A linkage to the 3′-hydroxyl group is shown.
All known transfer RNA molecules have the following features:
1. Each is a single chain containing between 73 and 93 ribonucleotides (~25 kDa).
2. The molecule is L-shaped (Figure 30.4).
3. They contain many unusual bases, typically between 7 and 15 per molecule. Some of these bases are methylated or dimethylated derivatives of A, U, C, and G formed by enzymatic modification of a precursor tRNA. Some methylations prevent the formation of certain base pairs, thereby rendering such bases accessible for interactions with other bases. In addition, methylation imparts a hydrophobic character to some regions of tRNAs, which may be important for their interaction with synthetases and ribosomal proteins. Other modifications alter codon recognition, as will be described shortly.
4. About half the nucleotides in tRNAs are base-paired to form double helices. The four helical regions are arranged to form two apparently continuous segments of double helix. These segments are like A-form DNA, as expected for an RNA helix (Section 4.2). One helix, containing the 5′ and 3′ ends, runs horizontally in the model shown in Figure 30.5. The other helix, which contains the anticodon and runs vertically in Figure 30.5, forms the other arm of the L.
Five groups of bases are not base-paired in this way: the 3′ CCA terminal region, which is part of a region called the acceptor stem; the TψC loop, which acquired its name from the sequence ribothymine-pseudouracilcytosine; the “extra arm,” which contains a variable number of residues; the DHU loop, which contains several dihydrouracil residues; and the anticodon loop. Most of the bases in the nonhelical regions participate in hydrogen-bonding interactions, even if the interactions are not like those in Watson–Crick base pairs. The structural diversity generated by this combination of helices and loops containing modified bases ensures that the tRNAs can be uniquely distinguished, though they are structurally similar overall.
5. The 5′ end of a tRNA is phosphorylated. The 5′ terminal residue is usually pG.
6. An activated amino acid is attached to a hydroxyl group of the adenosine residue in the amino acid-attachment site, located at the end of the 3′ CCA component of the acceptor stem (Figure 30.6). This single-stranded region can change conformation in the course of amino acid activation and protein synthesis.
7. The anticodon loop, which is present in a loop near the center of the sequence, is at the other end of the L, making accessible the three bases that constitute the anticodon.
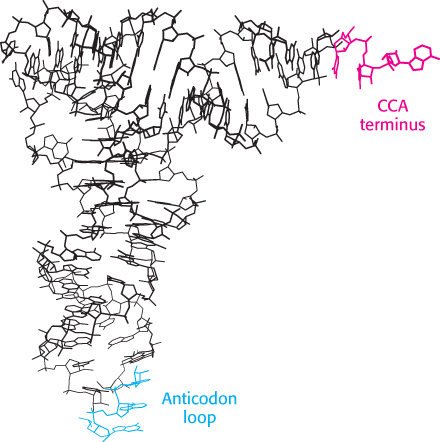
Figure 30.4:  Transfer RNA structure. Notice the L-shaped structure revealed by this skeletal model of yeast phenylalanyl-tRNA. The CCA region is at the end of one arm, and the anticodon loop is at the end of the other.
Transfer RNA structure. Notice the L-shaped structure revealed by this skeletal model of yeast phenylalanyl-tRNA. The CCA region is at the end of one arm, and the anticodon loop is at the end of the other.
[Drawn from 1EHZ.pdb.]
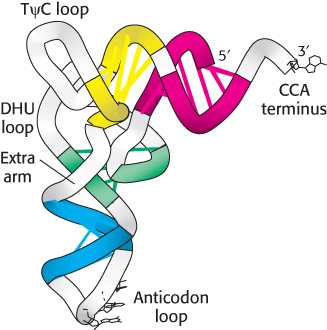
Figure 30.5:
Helix stacking in tRNA. The four double-stranded regions of the tRNA (Figure 30.3) stack to form an L-shaped structure.
[Drawn from 1EHZ.pdb.]
Thus, the architecture of the tRNA molecule is well suited to its role as adaptor: the anticodon is available to interact with an appropriate codon on mRNA while the end that is linked to an activated amino acid is well positioned to participate in peptide-bond formation.
Some transfer RNA molecules recognize more than one codon because of wobble in base-pairing
What are the rules that govern the recognition of a codon by the anticodon of a tRNA? A simple hypothesis is that each of the bases of the codon forms a Watson–Crick type of base pair with a complementary base on the anticodon of the tRNA. The codon and anticodon would then be lined up in an antiparallel fashion. In the diagram in the margin, the prime denotes the complementary base. Thus X and X′ would be either A and U (or U and A) or G and C (or C and G). According to this model, a particular anticodon can recognize only one codon.
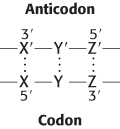
The facts are otherwise. As found experimentally, some tRNA molecules can recognize more than one codon. For example, the yeast alanyl-tRNA binds to three codons: GCU, GCC, and GCA. The first two bases of these codons are the same, whereas the third is different. Could it be that recognition of the third base of a codon is sometimes less discriminating than recognition of the other two? The pattern of degeneracy of the genetic code indicates that it might be so. XYU and XYC always encode the same amino acid; XYA and XYG usually do. These data suggest that the steric criteria might be less stringent for pairing of the third base than for the other two. In other words, there is some steric freedom (“wobble”) in the pairing of the third base of the codon.
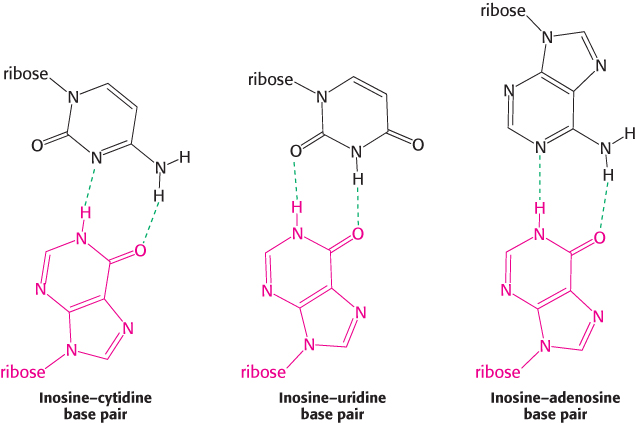
The wobble hypothesis is now firmly established (Table 30.2). The anticodons of tRNAs of known sequence bind to the codons predicted by this hypothesis. For example, the anticodon of yeast alanyl-tRNA is IGC. This tRNA recognizes the codons GCU, GCC, and GCA. Recall that, by convention, nucleotide sequences are written in the 5′ → 3′ direction unless otherwise noted. Hence, I (the 5′ base of this anticodon) pairs with U, C, or A (the 3′ base of the codon), as predicted.
Table 30.2: Allowed pairings at the third base of the codon according to the wobble hypothesis
Two generalizations concerning the codon–anticodon interaction can be made:
1. The first two bases of a codon pair in the standard way. Recognition is precise. Hence, codons that differ in either of their first two bases must be recognized by different tRNAs. For example, both UUA and CUA encode leucine but are read by different tRNAs.
2. The first base of an anticodon determines whether a particular tRNA molecule reads one, two, or three kinds of codons: C or A (one codon), U or G (two codons), or I (three codons). Thus, part of the degeneracy of the genetic code arises from imprecision (wobble) in the pairing of the third base of the codon with the first base of the anticodon. We see here a strong reason for the frequent appearance of inosine, one of the unusual nucleosides, in anticodons. Inosine maximizes the number of codons that can be read by a particular tRNA molecule. The inosine bases in tRNA are formed by the deamination of adenosine after the synthesis of the primary transcript.
Why is wobble tolerated in the third position of the codon but not in the first two? This question is answered by considering the interaction of the tRNA with the ribosome. As we will see, ribosomes are huge RNA–protein complexes consisting of two subunits, the 30S and 50S subunits. The 30S subunit has an RNA molecule, the 16S rRNA, that has three universally conserved bases—adenine 1492, adenine 1493, and guanine 530—that form hydrogen bonds on the minor-groove side but only with correctly formed base pairs of the codon–anticodon duplex (Figure 30.7). These interactions check whether Watson–Crick base pairs are present in the first two positions of the codon–anticodon duplex, but not the third position, so more-varied base pairs are tolerated. Thus, the ribosome plays an active role in decoding the codon–anticodon interactions.
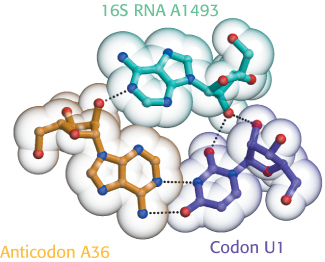
Figure 30.7: 16S rRNA monitors base-pairing between the codon and the anticodon. Adenine 1493, one of three universally conserved bases in 16S rRNA, forms hydrogen bonds with the bases in both the codon and the anticodon only if the codon and anticodon are correctly paired.
[Information from J. M. Ogle and V. Ramakrishnan, Annu. Rev. Biochem. 74:129–177, 2005, Fig. 2a.]