34.2 Antibodies Bind Specific Molecules Through Hypervariable Loops
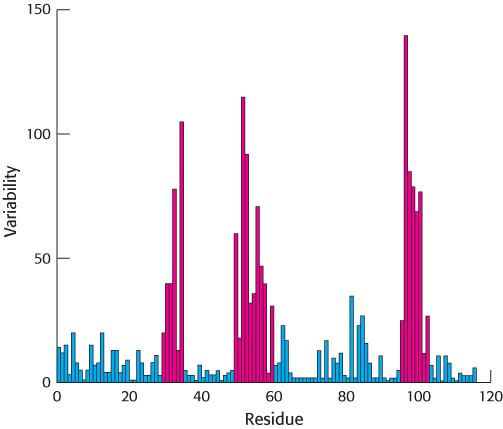
Figure 34.10: Immunoglobulin sequence diversity. A plot of sequence variability as a function of position along the sequence of the amino-terminal immunoglobulin domain of the H chain of human IgG molecules. Three regions (in red) show remarkably high levels of variability. These hypervariable regions correspond to three loops in the immunoglobulin domain structure.
[Data from R. A. Goldsby, T. J. Kindt, and B. A. Osborne, Kuby Immunology, 4th ed. (W. H. Freeman and Company, 2000), p. 91.]
A comparison of the amino acid sequences of different IgG antibodies from human beings or mice shows that the carboxyl-terminal half of the L chains and the carboxyl-terminal three-quarters of the H chains are very similar in all of the antibodies. Importantly, the amino-terminal domain of each chain is more variable, including three stretches of approximately 7 to 12 amino acids within each chain that are hypervariable, as shown for the H chain in Figure 34.10. The amino-terminal immunoglobulin domain of each chain is thus referred to as the variable region, whereas the remaining immunoglobulin domains are much more similar in all antibodies and are referred to as constant regions (Figure 34.11).
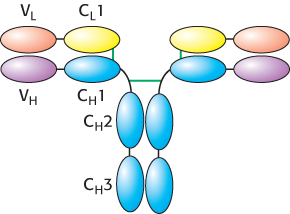
Figure 34.11: Variable and constant regions. Each L and H chain includes one immunoglobulin domain at its amino terminus that is quite variable from one antibody to another. These domains are referred to as VL and VH. The remaining domains are more constant from one antibody to another and are referred to as constant domains (CL1, CH1, CH2, and CH3).
The immunoglobulin fold consists of a beta-sandwich framework with hypervariable loops
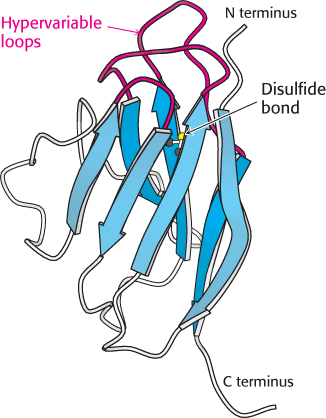
Figure 34.12:  Immunoglobulin fold. An immunoglobulin domain consists of a pair of β sheets linked by a disulfide bond and hydrophobic interactions. Notice that three hypervariable loops lie at one end of the structure.
Immunoglobulin fold. An immunoglobulin domain consists of a pair of β sheets linked by a disulfide bond and hydrophobic interactions. Notice that three hypervariable loops lie at one end of the structure.
[Drawn from 1DQJ.pdb.]
An IgG molecule consists of a total of 12 immunoglobulin domains. These domains have many sequence features in common and adopt a common structure, the immunoglobulin fold (Figure 34.12). Remarkably, this same structural domain is found in many other proteins that play key roles in both immune and nonimmune functions.
The immunoglobulin fold consists of a pair of β sheets, each built of antiparallel β strands, that surround a central hydrophobic core. A single disulfide bond bridges the two sheets. Two aspects of this structure are particularly important for its function. First, three loops present at one end of the structure form a potential binding surface. These loops contain the hypervariable sequences present in antibodies and in T-cell receptors. Variation of the amino acid sequences of these loops provides the major mechanism for the generation of the vastly diverse set of antibodies and T-cell receptors expressed by the immune system. These loops are referred to as hypervariable loops or complementarity-determining regions (CDRs). Second, the amino terminus and the carboxyl terminus are at opposite ends of the structure, which allows structural domains to be strung together to form chains, as in the L and H chains of antibodies. Such chains are present in several other key molecules in the immune system.
 The immunoglobulin fold is one of the most prevalent domains encoded by the human genome: more than 750 genes encode proteins with at least one immunoglobulin fold recognizable at the level of amino acid sequence. Such domains are also common in other multicellular animals such as flies and nematodes. However, from inspection of amino acid sequence alone, immunoglobulin-fold domains do not appear to be present in yeast or plants, although these organisms possess other structurally similar domains, including the key photosynthetic electron-transport protein plastocyanin in plants (Section 19.3). Thus, the immunoglobulin-fold family appears to have expanded greatly along evolutionary branches leading to animals—particularly, vertebrates.
The immunoglobulin fold is one of the most prevalent domains encoded by the human genome: more than 750 genes encode proteins with at least one immunoglobulin fold recognizable at the level of amino acid sequence. Such domains are also common in other multicellular animals such as flies and nematodes. However, from inspection of amino acid sequence alone, immunoglobulin-fold domains do not appear to be present in yeast or plants, although these organisms possess other structurally similar domains, including the key photosynthetic electron-transport protein plastocyanin in plants (Section 19.3). Thus, the immunoglobulin-fold family appears to have expanded greatly along evolutionary branches leading to animals—particularly, vertebrates.
X-ray analyses have revealed how antibodies bind antigens
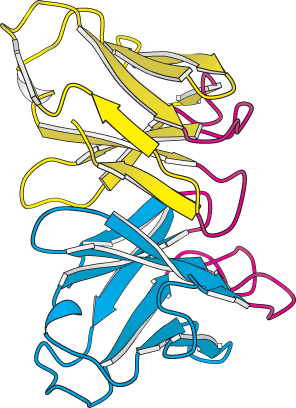
Figure 34.13:
Variable domains. A side view of the variable domains of the L chain (yellow) and the H chain (blue); the complementarity-determining regions (CDRs) are shown in red. Notice that the six CDRs come together to form a binding surface. The specificity of the surface is determined by the sequences and structures of the CDRs.
[Drawn from 1DQJ.pdb.]
For each class of antibody, the variable domains at the amino-terminal ends of the L and H chains (designated VL and VH) come together to form a binding surface. The positions of the CDRs are striking. These hypervariable sequences, present in three loops of each domain, come together so that all six loops form a single surface at the end of each arm (Figure 34.13). Because virtually any VL can pair with any VH, a very large number of different binding sites can be constructed by their combinatorial association.
The results of x-ray crystallographic studies of several hundred large and small antigens bound to Fab molecules have been sources of much insight into the structural basis of antibody specificity. The binding of antigens to antibodies is governed by the same principles that govern the binding of substrates to enzymes. The interaction between complementary shapes results in numerous contacts between amino acids at the binding surfaces of both molecules. Many hydrogen bonds, electrostatic interactions, and van der Waals interactions, reinforced by hydrophobic interactions, combine to give specific and strong binding.
A few aspects of antibody binding merit specific attention, inasmuch as they relate directly to the structure of immunoglobulins. The binding site on the antibody incorporates some or all of the CDRs in the variable domains of the antibody. Small molecules are likely to make contact with fewer CDRs, with perhaps 15 residues of the antibody participating in the binding interaction. Macromolecules often make more extensive contact, sometimes interacting with all six CDRs and 20 or more residues of the antibody. Small molecules often bind in a cleft of the antigen-binding region. Macromolecules, such as globular proteins, tend to interact across larger, fairly flat apposed surfaces bearing complementary protrusions and depressions.
A well-studied case of small-molecule binding is seen in an example of phosphorylcholine bound to Fab. Crystallographic analysis revealed phosphorylcholine bound to a cavity lined by residues from five CDRs—two from the L chain and three from the H chain (Figure 34.14). The positively charged trimethylammonium group of phosphorylcholine is buried inside the wedge-shaped cavity, where it interacts electrostatically with two negatively charged residues, a glutamate and an aspartate. The negatively charged phosphoryl group of phosphorylcholine binds to the positively charged guanidinium group of an arginine residue at the mouth of the crevice and is hydrogen bonded to the side chain of a nearby tyrosine residue. Numerous van der Waals interactions, such as those made by a tryptophan side chain, also stabilize this complex.
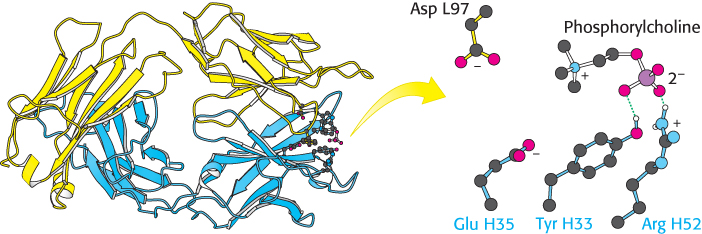
Figure 34.14: Binding of a small antigen. The structure of a complex between an Fab fragment of an antibody (blue and yellow) and its target—in this case, phosphorylcholine. Notice that residues from the antibody interact with phosphorylcholine through hydrogen bonding and electrostatic interactions.
[Drawn from 2MCP.pdb.]
Residues from five CDRs participate in the binding of phosphorylcholine to human Fab. This binding does not significantly change the structure of the antibody, yet induced fit plays a role in the formation of many antibody–antigen complexes. A malleable binding site can accommodate many more kinds of ligands than can a rigid one. Thus, induced fit increases the repertoire of antibody specificities.
Large antigens bind antibodies with numerous interactions
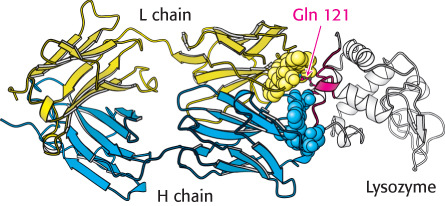
Figure 34.16:
Antibody–protein interactions. Notice that a single residue of lysozyme, glutamine 121, penetrates more deeply into the antibody binding site.
[Drawn from 1FDL.pdb.]
How do large antigens interact with antibodies? A large collection of antibodies against hen egg-white lysozyme has been structurally characterized in great detail (Figure 34.15). Each different antibody binds to a distinct surface of lysozyme. Let us examine the interactions in one of these complexes (complex ii in Figure 34.15) in detail. This antibody binds two polypeptide segments of lysozyme that are widely separated in the primary structure (Figure 34.16).
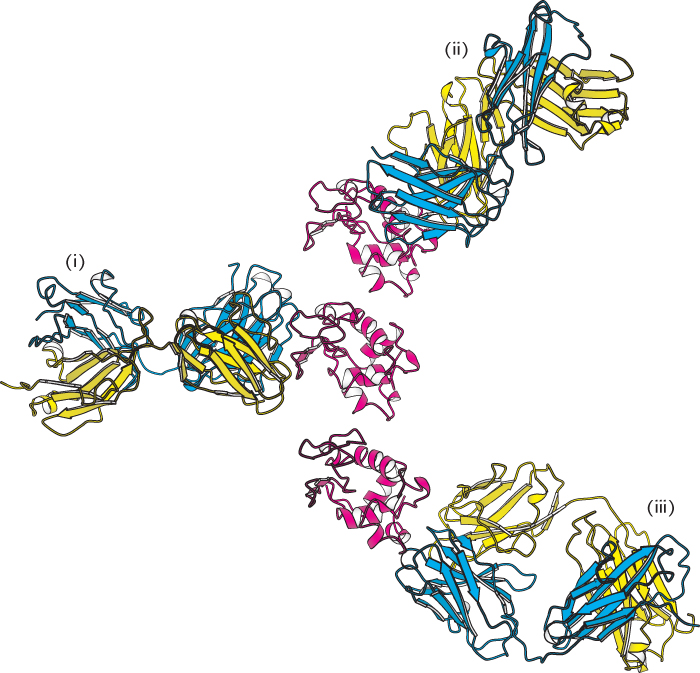
Figure 34.15:
Antibodies against lysozyme. The structures of three complexes (i, ii, iii) between Fab fragments (blue and yellow) and hen egg-white lysozyme (red) shown with lysozyme in the same orientation in each case. Notice that the three antibodies recognize completely different epitopes on the lysozyme molecule.
[Drawn from 3HFL, 1DQJ, and 1FDL.pdb.]
All six CDRs of the antibody make contact with this epitope. The region of contact is quite extensive (about 30 × 20 Å). The apposed surfaces are rather flat. The only exception is the side chain of glutamine 121 of lysozyme, which penetrates deeply into the antibody’s binding site, where it forms a hydrogen bond with a main-chain carbonyl oxygen atom and is surrounded by three aromatic side chains. The formation of 12 hydrogen bonds and numerous van der Waals interactions contributes to the high affinity (Kd = 20 nM) of this antibody–antigen interaction. Examination of the Fab molecule without bound protein reveals that the structures of the VL and VH domains change little on binding, although they slide 1 Å apart to allow more intimate contact with lysozyme.