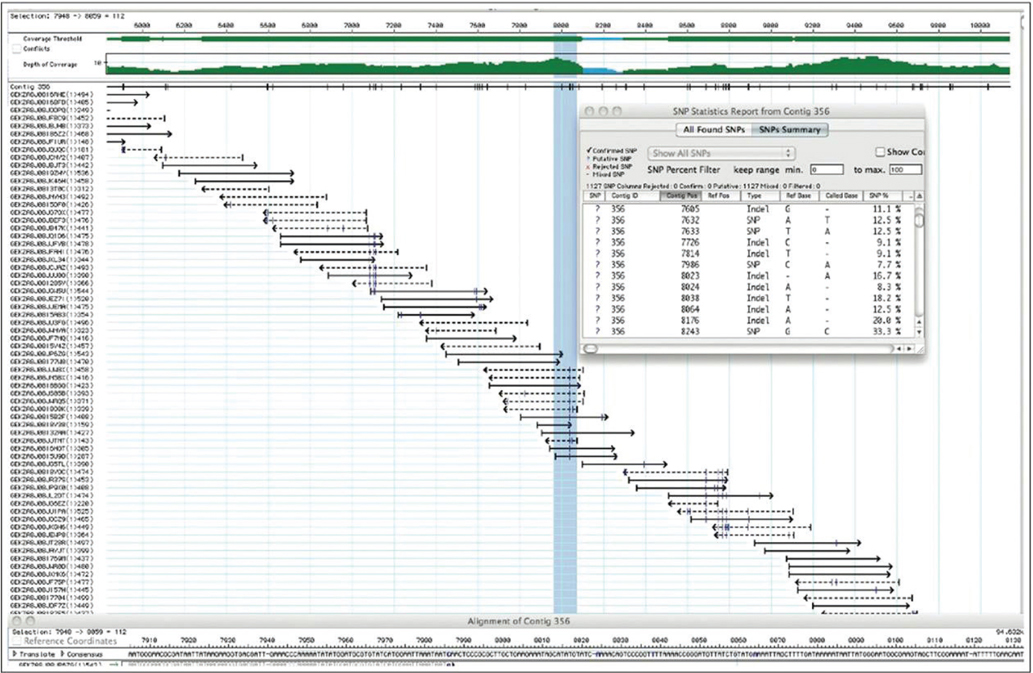
Sequence assembly. In a genomic sequence, each base pair of the genome is usually represented in many of the sequenced fragments, referred to as reads. Shown is a small part of the sequence of a new variant species of E. coli, with the reads generated by a 454 sequencer. The numbers at the top represent genomic base- r-