8.1 GENOMES AND GENOMICS
The word “genome,” coined by German botanist Hans Winkler in 1920, was derived simply by combining gene and the final syllable of chromosome. An organism’s genome is defined today as the complete haploid genetic complement of a typical cell. In essence, a genome is one copy of the hereditary information required to specify the organism. For sexually reproducing organisms, the genome includes one set of autosomes and one of each type of sex chromosome. When cells have organelles that also contain DNA, the genetic content of the organelles is not considered part of the nuclear genome. Mitochondria are found in all eukaryotic cells, and chloroplasts occur in the light-harvesting cells of photosynthetic organisms. Each of these organelles has its own distinct genome. In viruses, which can have genetic material composed of either DNA or RNA, the genome is a complete copy of the nucleic acid required to specify the virus.
In diploid organisms, sequence variations exist between the two copies of each chromosome present in a cell. Subtle as they are (estimates range from 0.1% to 0.5% variance), these differences are used to solve crimes (see Highlight 7-1), define parentage, and help trace the path of an inherited disease through generations of an affected family. As sequencing methods become more sophisticated, sequences of the complete genetic complement of a diploid cell—diploid genomes—are beginning to appear.
The study of complete genomes was rudimentary until the advent of genome sequencing projects in the 1980s. In 1986, Thomas H. Roderick of the Jackson Laboratories in Bar Harbor, Maine, came up with Genomics as the name for a new journal, and the word ended up defining a new field. The modern science of genomics is dedicated to the study of DNA on a cellular scale. Advances in this field have been propelled by improvements in sequencing technology (described in Chapter 7), in computer technologies, and in innovative approaches to the organization and searching of stored information.
Many Genomes Have Been Sequenced in Their Entirety
The genome is the ultimate source of genetic information about an organism. Less than 10 years after the development of practical DNA sequencing methods, serious discussions began about the prospects for sequencing the entire 3 × 109 base pairs (bp) of the human genome. Although the effort was inspired by our natural curiosity about ourselves, it has become much more than a human genome project. Genome sequencing in the twenty-first century is becoming routine. The number of genomes sequenced in their entirety is now in the tens of thousands and includes organisms ranging from bacteria to mammals.
The International Human Genome Project got under way in the late 1980s. Several additional and closely linked projects focused on organisms other than humans. The first complete genome to be sequenced was that of the bacterium Haemophilus influenzae, in 1995 (see the How We Know section at the end of this chapter). The first eukaryotic genome sequenced was that of the yeast Saccharomyces cerevisiae, in 1996, and the genome sequence for the bacterium Escherichia coli became available in 1997 (Figure 8-1). The much larger effort directed at the human genome was also accelerating.
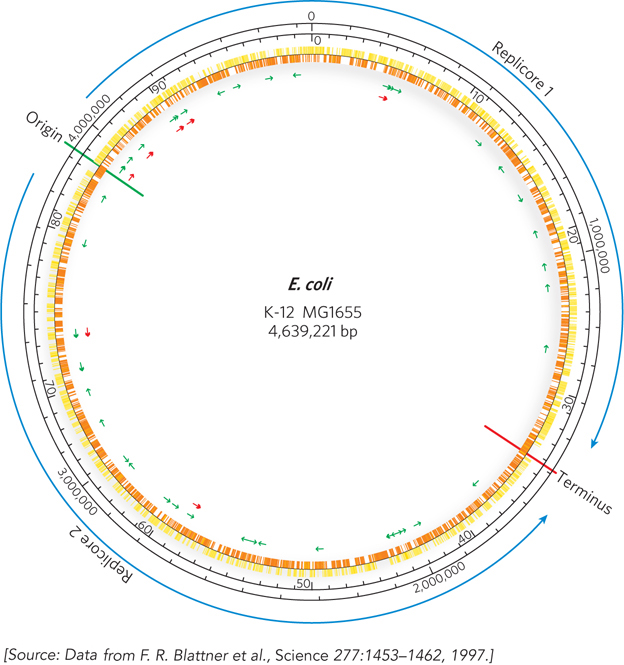
Figure 8-1: A snapshot of the Escherichia coli genome. K-12 MG1655 is the most commonly used laboratory strain of E. coli. The origin and terminus of replication are shown as green and red lines, respectively. Replication proceeds in two directions from the origin, dividing the genome into two regions, or replicores, which are replicated separately. The two outer black circles provide genome reference points in terms of number of base pairs (outer) and minutes (inner). The use of 100 minutes as a genomic yardstick in this chromosome is derived from the approximately 100 minutes it takes to transfer the E. coli chromosome from donor cell to recipient during bacterial conjugation. Yellow and orange markings on the inner circle denote protein-coding genes. Green and red arrows indicate the locations of genes for tRNA and rRNA, respectively.

Francis Collins
The Human Genome Project eventually included significant contributions from 20 sequencing centers distributed among six nations: the United States, Great Britain, Japan, France, China, and Germany. Some general coordination was provided by the Office of Genome Research at the National Institutes of Health in the United States, led first by James Watson and after 1992 by Francis Collins. However, much of the effort relied on the informal and very successful international collaborations.
The published sequence of the human genome is actually a composite, derived from several anonymous donors. Although the DNA of several individuals is represented, the sequence in any given genomic region is generally from one individual. Research teams used restriction enzymes to partially digest the entire human genome, and then cloned suitably long segments into BAC and YAC vectors (bacterial and yeast artificial chromosomes are discussed in Chapter 7). Overlapping clones in the resulting libraries were identified by hybridization and other methods and organized into long contiguous stretches of chromosomal DNA called contigs. Each contig included at least one and usually many identifying sequences that had already been mapped to a particular region of a particular chromosome. These regions were either a unique and previously characterized sequence called a sequence tagged site (STS) or a gene for which expression could be monitored, known as an expressed sequence tag (EST). The STS and EST landmarks in the contigs were often sequences that had been roughly mapped along a specific chromosome. These landmark-containing contigs could thus be ordered along each chromosome, gradually defining a physical map of the genome (Figure 8-2).
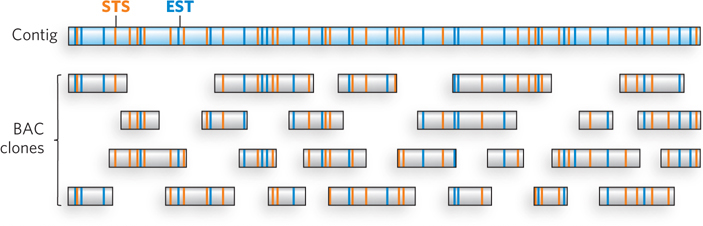
Figure 8-2: Mapping BACs into a contig. A contig of a chromosomal region is constructed by identifying the sequence tagged site (STS; orange) and expressed sequence tag (EST; blue) markers in each BAC and mapping them to the contig.
The contigs were then divided up between the international sequencing centers, and each center began sequencing the mapped BAC or YAC clones corresponding to its assigned segments of the genome. Because many of the clones were more than 100,000 bp long, and contemporary sequencing techniques resolved only 600 to 750 bp of sequence at a time, each clone had to be sequenced in pieces. The strategy was a shotgun approach, in which researchers used powerful new automated sequencers to sequence random segments of a given BAC or YAC clone, then assembled the entire clone by computerized identification of overlaps. Each clone was sequenced at least four to six times to ensure accuracy. The data were made available in the growing genome database.

J. Craig Venter
A competing commercial effort to sequence the human genome had been initiated by the newly established Celera Corporation in 1997. Led by J. Craig Venter, the Celera group made use of a different strategy, called whole-genome shotgun sequencing, which eliminates the step of assembling a physical map of the genome. Instead, teams sequenced DNA segments from throughout the genome, at random. The sequenced segments were ordered by the computerized identification of sequence overlaps (with some reference to the public project’s detailed and published physical map). Like the public genome project, the Celera effort used DNA from several human donors. About 70% of the sequence comes from one male donor—Craig Venter himself.
At the outset of the Human Genome Project in the 1980s, shotgun sequencing on this scale had been deemed impractical because the sequence assembly was too computationally complex. However, by 1997, advances in computer software and sequencing automation had made the approach feasible. The ensuing race between private and public efforts substantially shortened the timeline for completing the project (Figure 8-3). After publication of the draft human genome sequence in February 2001, two years of follow-up work ensued, to eliminate nearly a thousand discontinuities and to provide high-quality sequence data, contiguous throughout the genome. The project was completed in April 2003, several years ahead of schedule.

Figure 8-3: The genome sequencing timeline. Preparatory work for the Human Genome Project, including extensive mapping to provide genome landmarks, occupied much of the 1990s. The rapid development of sequencing methods and strategies initiated a broad range of additional sequencing efforts. By the time the draft human genome was published in 2001, hundreds of other projects were already under way. Many genome projects involving species that are widely used in research have their own websites, which serve as central repositories for the most recent data. The increasing inclusion of species that are close human relatives and recent human ancestors is both clarifying human evolution and providing important resources for the discovery of genes involved in human diseases.
The human genome is only part of the genome sequencing story, however, and an increasingly small part. The genomes of many other species have been sequenced in the continuing effort, gradually providing a unique look at genomic complexity throughout the three domains of living organisms: Bacteria, Archaea, and Eukarya (eukaryotes) (see Figure 8-3). Whereas many early sequencing projects focused on species commonly used in research laboratories, the programs have expanded to include a wide range of species of practical, medical, agricultural, and evolutionary interest. Completed bacterial genomes include at least one species from virtually every known bacterial family, and completed eukaryotic genomes number in the thousands. Each completed genome becomes a resource for scientists around the world who use that organism in their research, facilitating the identification of important genes. Thousands of individual human genomes have been sequenced, and the number is growing exponentially as genome-based, personalized medicine becomes a reality. The sequencing efforts have even expanded to include extinct species such as Homo neanderthalensis, as well as humans who died in past millennia. The many genome sequences provide a source for broad comparisons that help pinpoint both variable and highly conserved gene segments and allow the identification of genes that are unique to a species or groups of species. Efforts to map genes, identify new proteins and disease genes, elucidate genetic patterns of medical interest, and trace our evolutionary history, as well as many other initiatives, are under way.
Annotation Provides a Description of the Genome
A genome sequence is simply a very long string of A, G, T, and C residues. The value of this sequence information depends almost entirely on the manner in which the information is organized when it is stored. The critical process of genome annotation yields a listing of information about the location and function of genes and other critical sequences. Genome annotation converts the sequence itself to information that any researcher can use. Much of the effort focuses on genes encoding RNA and protein, because such genes are most often the target of scientific investigations. Every newly sequenced genome includes many genes—often 40% or more of the total—about which little or nothing is known. Here, the annotation exercise is most challenging.
Protein and RNA function can be described on three levels. Phenotypic function describes the effects of a gene product on the entire organism. For example, the loss or mutation of a particular protein may lead to slower growth, altered development, or even death. Cellular function is a description of the metabolic processes in which a gene product participates and of the interactions of that gene product with other proteins or RNAs in the cell. Molecular function refers to the precise biochemical activity of a protein or an RNA, such as the reactions an enzyme catalyzes, the ligands a receptor binds, or the complex formed between a specific RNA and a protein. Each of these functions can be elucidated by computational and experimental approaches, some of which are described here. Additional techniques are presented in Section 8.2.
Computational approaches involve Web-based programs that are used to define gene locations and assign tentative gene functions (where possible), based on similarity to genes previously studied in other genomes. Most of these programs are freely available on the Internet, although some are written for specific purposes by individual researchers. For investigating the function of a particular gene, resources such as the classic BLAST (Basic Local Alignment Search Tool) algorithm allow a rapid search of all genome databases for sequences related to one that a researcher has just generated. Two other prominent Internet resources are the NCBI (National Center for Biotechnology Information) site, sponsored by the National Institutes of Health, and the Ensembl site, cosponsored by the EMBL-EBI (European Molecular Biology Laboratory–European Bioinformatics Institute) and the Wellcome Trust Sanger Institute.
The availability of many genome sequences in online databases enables researchers to assign gene functions by genome comparisons, an enterprise referred to as comparative genomics. Sequence comparison can be done with DNA, RNA, or protein. Any two genes with a demonstrable sequence similarity, whether or not they are closely related by function, are called homologs. The sequence similarity (homology) implies an evolutionary relationship. Quite often, sequence similarity and a functional relationship go hand in hand. When two genes in different species possess a clear sequence and functional relationship to each other, they are known as orthologs—genes derived from an ancestral gene in the last common ancestor of these two species. Paralogs are genes that are similarly related to each other but within a single species; they arise most often from gene duplication in a single genome, followed by specialization of one or both copies of the gene over the course of evolution. If the function of any gene has been characterized for one species, this information can be used to at least tentatively assign gene function to a related gene in a second species. The entire process of annotation relies heavily on evolutionary theory. Comparing genomes from different species is productive precisely because all organisms share a common ancestor.
Gene identity is often easiest to discern when comparing genomes from closely related species, such as mice and humans, although many clearly orthologous genes have been identified in species as distant as bacteria and humans. In many cases, even the order of genes on a chromosome is conserved over large segments of the genomes of closely related species. Conserved gene order, or synteny, provides additional evidence for an orthologous relationship between genes at identical locations in the related segments (Figure 8-4). The distinction between orthologs and paralogs was introduced by Walter Fitch in 1970, and its importance was established with the advent of genome sequencing projects in the 1990s. As the number of known genome sequences increases, many genes and genomic segments can be productively annotated by using automated tools available on the NCBI and Ensembl websites.
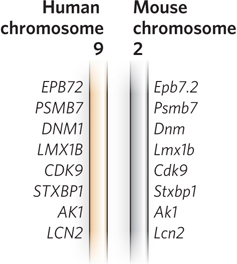
Figure 8-4: Synteny in the human and mouse genomes. Large segments of the two genomes have closely related genes aligned in the same order on the chromosomes. In these short segments of human chromosome 9 and mouse chromosome 2, the genes exhibit a very high degree of homology, as well as the same gene order. The different lettering schemes for the gene names simply reflect the different naming conventions for the two species.
In every newly described genome sequence, the many genomic segments and genes that have never been characterized—that unknown 40% or so of the total—represent a special challenge. Elucidation of gene function in these cases will probably take many decades. Some experimental approaches exist, and new ones are being developed. Many of the current approaches again focus on protein-coding genes; for several genomes, such as those of S. cerevisiae and the plant Arabidopsis thaliana, gene knockout (inactivation) collections have been developed by genetic engineering. Each strain in an organism’s collection has a different inactivated gene, and a high fraction of genomic genes (excluding a core of essential genes required for life at all times) are represented. If the growth patterns or other properties of the organism change when the gene is inactivated, this provides information on the phenotypic function of the protein product of the gene. In other available libraries, each gene in a specific genome is expressed as a tagged fusion protein (see Chapter 7). The tags may be designed to allow isolation of the protein, investigate interactions with other proteins, or explore subcellular localization. Some approaches for using tags to determine the function of genes are described in more detail in Section 8.2.
Genome Databases Provide Information about Every Type of Organism
The available genome sequences are helpful to research in all biological disciplines. Increasingly, they are inspiring molecular biologists to ask questions that, until now, could not be answered. Just a brief overview will illustrate the utility of expanding the Human Genome Project to essentially all species.
Viruses Viruses are not free-living organisms but obligate intracellular parasites: thus, every virus is a pathogen of some organism. The viruses that are human pathogens—such as SARS (see this chapter’s Moment of Discovery)—are of special interest. However, viruses that infect farm animals, food crops, landscape plants, and many other organisms can be economically important. Bacteria also serve as hosts to viruses, which are generally termed bacteriophages. Even these are of medical interest, because bacteriophages that kill pathogenic bacteria have therapeutic potential. Viruses can be divided into seven classes, depending on whether the genomic nucleic acid is RNA or DNA, whether it is single-stranded or double-stranded, and the mechanisms employed to replicate it (Table 8-1). Viruses vary a great deal in genomic complexity, ranging from a mere 2,000 nucleotides (found in a few single-stranded DNA viruses that infect vertebrates) to around 1.2 million bp (in a double-stranded DNA virus that infects amoebas). The number of viral genomes that have been or are now being sequenced has grown so quickly that estimates become obsolete almost as soon as they are generated. The ongoing effort will greatly aid future progress in medicine and agriculture.
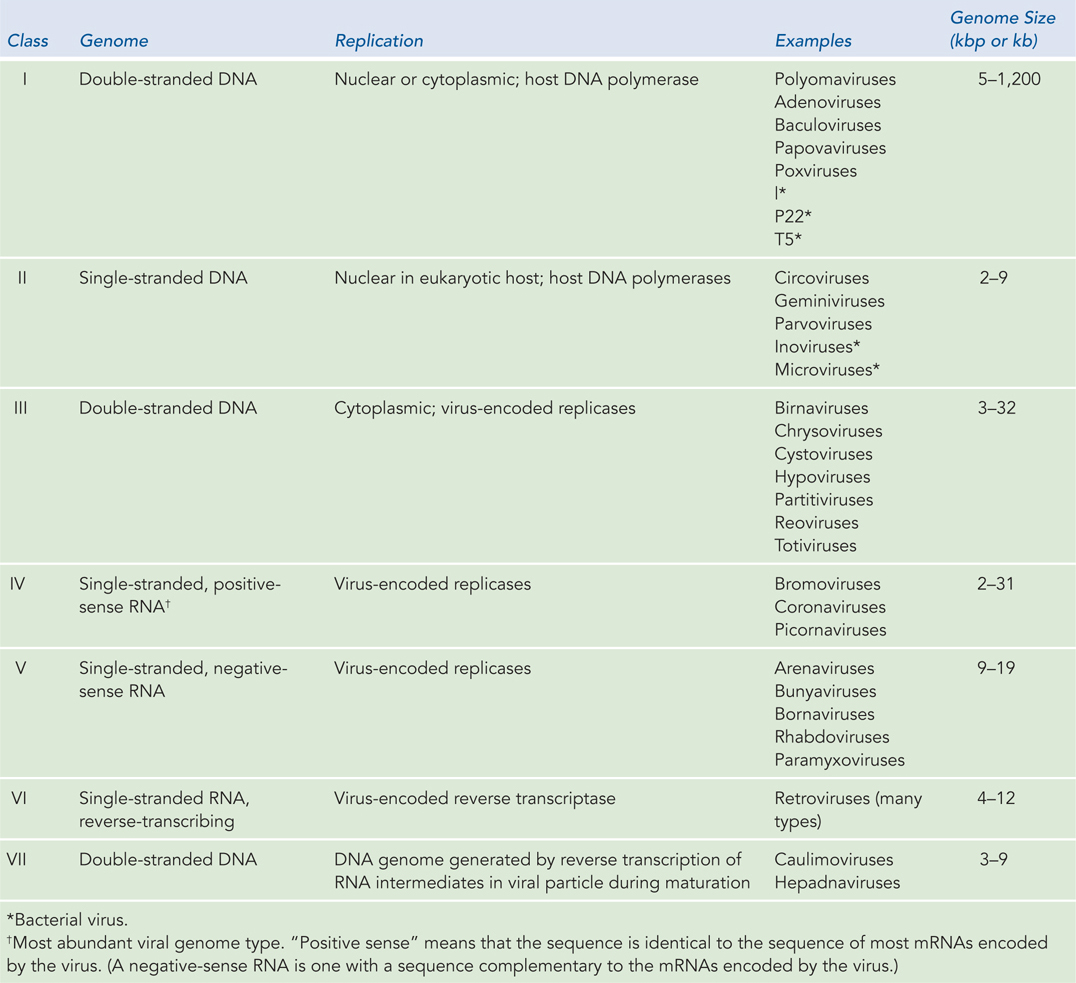
Figure 8-1: The Seven Classes of Viruses
Bacteria Bacteria inhabit every environment—from polar ice to deserts, from ocean depths to kitchen counters and the soil in your backyard. Some are pathogens. Others help digest our food, convert atmospheric nitrogen to forms that all organisms can use, convert carbon dioxide to oxygen, and carry out myriad other tasks without which all other life forms would perish. With that in mind, molecular biologists are subjecting thousands of representative bacterial species to genome sequencing.
In the past few decades, researchers have realized that a vast number of bacterial species remain uncharacterized. Many bacteria live in interdependent microbial communities and cannot be cultured in pure form in the laboratory. Examples are found in the human intestine, in the termite gut, and in the effluent of deep-sea steam vents. Many of these bacteria are important to human health, both directly and indirectly. Bacteria are so plentiful in humans—in our intestines, on our skin, in our saliva, and so on—that they represent 1% to 3% of the mass of a typical person, with symbiotic bacterial cells outnumbering their human host cells by a factor of 10 to 1. Other bacteria are of economic importance. For instance, an understanding of the microbial processes that allow termites to digest cellulose in their gut could provide new ways to convert grass and other cellulose materials to usable fuels.
The need to know more about these microbial communities has given rise to a subdiscipline of genomics: metagenomics. In metagenomics projects, DNA is isolated not from a single bacterial species but from an entire community of microbial species (Highlight 8-1). The DNA is sequenced by a shotgun technique, and the researcher uses computer programs to assemble overlapping segments derived from individual genomes.
HIGHLIGHT 8-1 TECHNOLOGY: Sampling Biodiversity with Metagenomics
Most of the biological diversity on our planet is found in microorganisms. However, we know surprisingly little about Earth’s microbial diversity. A wealth of microbial diversity remains to be discovered in the world’s swamps, deserts, and oceans, involving species that cannot yet be cultured. Assessing the diversity in communities of microorganisms is one goal of the new discipline of metagenomics.
The sampling involves shotgun DNA sequencing on a truly grand scale. Individual species are not isolated. Instead, an entire microbial population is taken from a given environment, and DNA sequences from that population are analyzed at random. Early approaches have looked at a biofilm in an acid mine, soil in Minnesota, water samples from the Sargasso Sea, whale falls (whales that have died and sunk to the ocean floor), and human feces. DNA from the bacteria and/or viruses in the sample is broken into fragments and sequenced at random. Computerized analyses identify any overlapping sequences and link them into longer contigs. These genomic snippets are assembled in a database. The diversity can be measured by focusing on specific genes. For example, the 16S rRNA gene is universal in bacteria and is often used as a benchmark for defining species.
This technology has given rise to some very ambitious metagenomics initiatives. One was carried out by Craig Venter and his coworkers at the J. Craig Venter Institute (Rockville, Maryland) in the spring of 2003. A 30-meter sailing sloop, the Sorcerer II, was converted into an oceanic research vessel to carry out the Global Ocean Sampling (GOS) expedition. Launched in March 2004 in Halifax, Canada, the nearly two-year research voyage circumnavigated the globe (Figure 1). Samples of ocean water were taken every 200 nautical miles. Microorganisms were strained from the water with a series of filters and sent back to the lab for extraction of DNA and sequencing. The result is the largest database of DNA sequences derived from marine organisms yet released into the public domain. More than 7.7 million sequences are included in the GOS database, encompassing more than 6.3 billion bp of DNA and representing more than 800 species—perhaps half of which were previously unknown. Millions of new proteins have been discovered, and thousands of new protein families. The voyages continue.
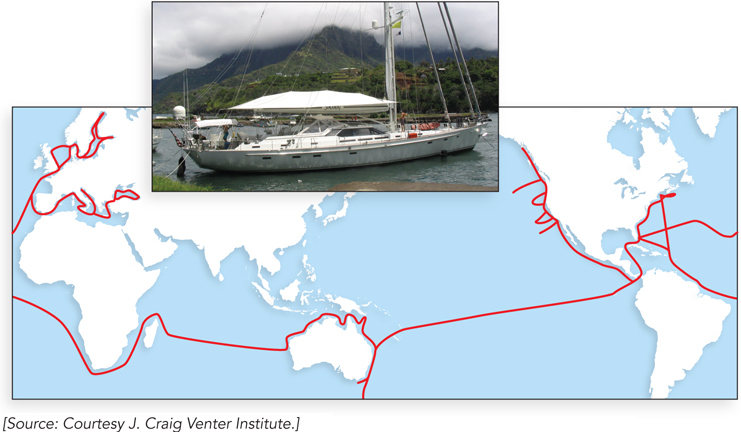
FIGURE 1 The route taken by Sorcerer II.
A second effort in metagenomics is to map the human microbiome. Major contributions come from the U.S. Human Microbiome Project of the National Institutes of Health and the European MetaHIT (Metagenomics of the Human Intestinal Tract) program, both launched in 2008. The goal is to define the bacteria and other microorganisms (including some archaea and yeasts) associated with humans. Perhaps even more important, the projects will determine how the microbiome changes in response to geography, diet, age, pregnancy, disease state, and many other factors. This effort is inspired in part by an increasing awareness of the role of the human microbiome in health. Clear links are becoming apparent between a disruption of homeostasis between microbiota and their human host (referred to as dysbiosis) and the development of disease states such as inflammatory bowel disease, obesity, and type 2 diabetes. Microbe replacement therapy could be in your future.
When the community includes only a handful of species, researchers can reconstruct multiple genomes from these mixed samples. The process has some unique complexities, many related to the close evolutionary relationship of many bacterial species. This results in similarities over large stretches of genomic nucleic acid that can complicate the computerized assembly of genomes from short sequence “reads.” Assembling genomes from microbial communities with hundreds of different species will require further advances in both sequencing technologies and assembly programs.
Archaea In 1977, Carl Woese and his colleagues introduced the world to a new domain of living organisms, the Archaebacteria, now renamed Archaea. A careful study of 16S ribosomal RNA sequences led to the discovery of this previously unsuspected group. The archaea (singular, archaeon) are single-celled organisms, very similar in appearance to bacteria, and like bacteria are ubiquitous. However, many of the most interesting species are extremophiles, inhabiting hot springs or water with very high salinity or other unusual environments. Sharing some properties with both bacteria and eukaryotes, archaea have nevertheless evolved as an independent line. Their contributions to the chemistry of the biosphere make them important targets for study and genome sequencing.
Eukaryotes Many eukaryotic genomes are considerably larger than genomes in the other two domains. Nevertheless, the sequencing of even very large eukaryotic genomes is becoming routine. Databases already contain complete genomes ranging from single-celled eukaryotes such as S. cerevisiae to nematodes, plants, insects, and mammals. Orthologs of genes involved in important processes and disease states in humans can almost always be found in the genomes of model organisms, facilitating laboratory research into gene function. Specialized databases have been developed for the genomes of organisms that are of particular interest to science, including mouse, fruit fly, mustard weed, and yeast (see the Model Organisms Appendix). Other databases are being established that focus on plant and animal species critical to agriculture, such as corn, rice, and cattle. Some databases focus on specific types of genes. All of these databases are easily found through an Internet search or via links on the NCBI and Ensembl websites. Individual human genome sequences are also available online, including those of James Watson and Craig Venter!
The Human Genome Contains Many Types of Sequences
All of these rapidly growing databases have the potential not only to fuel advances in biology but to change the way we think about ourselves. What does our own genome, and its comparison with those of other organisms, tell us?
In some ways, we are not as complicated as we once imagined. Decades-old estimates that humans had about 100,000 genes within the approximately 3.2 × 109 bp of the human genome have been supplanted by the discovery that we have only about 20,000 protein-coding genes—less than twice the number in a fruit fly (13,601 genes), not many more than in a nematode worm (19,735 genes), and fewer than in a rice plant (∼38,000 genes).
In other ways, however, we are more complex than we previously realized. The study of eukaryotic chromosome structure and, more recently, the sequencing of entire eukaryotic genomes have revealed that many, if not most, eukaryotic genes contain one or more intervening segments of DNA that do not code for the amino acid sequence of the polypeptide product. These nontranslated inserts interrupt the otherwise colinear relationship between the gene’s nucleotide sequence and the amino acid sequence of the encoded polypeptide. Such nontranslated DNA segments are called introns (or intervening sequences), and the coding segments are called exons (Figure 8-5). Few bacterial genes contain introns. The process of removing introns from a primary RNA transcript to generate a transcript that can be translated contiguously into a protein product is known as splicing (described in Chapter 16). An exon often (but not always) encodes a single domain of a larger, multidomain protein. Humans share many protein domain types with plants, worms, and flies, but use these domains in more complex arrangements. Alternative modes of gene expression and RNA splicing permit the production of alternative combinations of exons, leading to the production of more than one protein from a single gene. Humans and other vertebrates engage in this process far more than do bacteria, worms, or any other form of life—thereby allowing greater complexity in the proteins generated.
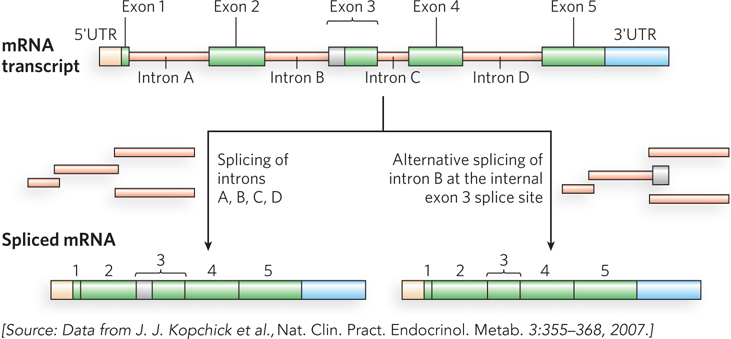
Figure 8-5: Introns and exons. The gene for human growth hormone 1 (GH1) contains five exons and four introns, along with 5′ and 3′ untranslated regions (5′UTR and 3′UTR). Two of the several alternative patterns of splicing are shown here. Alternative splicing allows cells to synthesize different variants of a protein from one gene.
In mammals and some other eukaryotes, the typical gene has a much higher proportion of intron DNA than exon DNA; in most cases, the function of introns is not clear. Less than 1.5% of human DNA is “coding” or exon DNA, carrying information for protein or RNA products (Figure 8-6a). However, when the much larger introns are included in the count, as much as 30% of the human genome consists of genes. Several efforts are under way to categorize the protein-coding genes by function (Figure 8-6b).
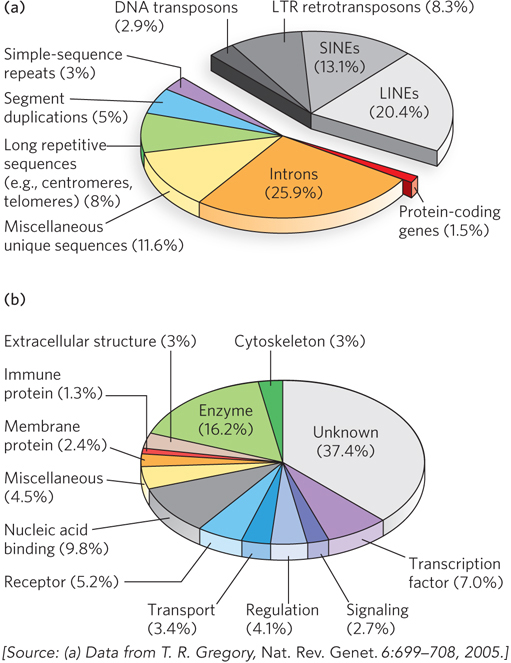
Figure 8-6: A snapshot of the human genome. (a) This pie chart shows the proportions of various types of sequences in the human genome. The classes of transposons that represent nearly half of the total genomic DNA are indicated in shades of gray. LTR retrotransposons are retrotransposons with long terminal repeats. Long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs) are special classes of particularly common DNA transposons (see Chapter 14). (b) The approximately 20,000 protein-coding genes in the human genome can be classified by the type of protein encoded.

Barbara McClintock, 1902–1992
The relative paucity of genes in the human genome leaves a lot of DNA unaccounted for. Much of the nongene DNA is in the form of repeated sequences of several kinds. Perhaps most surprising, about half the human genome is made up of moderately repeated sequences that are derived from transposable elements—segments of DNA, ranging from a few hundred to several thousand base pairs long, that can move from one location to another in the genome. Originally discovered in corn by Barbara McClintock, transposable elements, or transposons, are a kind of molecular parasite. They efficiently make their home in the genomes of essentially every organism. Many transposons contain genes encoding the proteins that catalyze the transposition process itself, as described in more detail in Chapter 14. There are multiple classes of transposons in the human genome. Many are strictly DNA segments, slowly increasing in number as a result of replication events coupled to the transposition process. Some, called retrotransposons, are closely related to retroviruses, transposing from one genomic location to another through RNA intermediates that are reconverted to DNA by reverse transcription. Some transposons in the human genome are active, moving at a low frequency, but most are inactive, evolutionary relics altered by mutations. Transposon movement can lead to the redistribution of other genomic sequences and has played a major role in human evolution.
Once the protein-coding genes (including exons and introns) and transposons are accounted for, perhaps 25% of the total DNA remains (the purple, blue, green, and yellow segments in Figure 8-6a). As a follow-up to the Human Genome Project, an initiative called ENCODE was launched by the National Human Genome Research Institute in 2003. Involving a worldwide consortium of research groups, its purpose is to identify functional elements in the human genome. As the project proceeds, the nongene DNA in the human genome is becoming less of a mystery. ENCODE has revealed that the vast majority (>80%, including most transposons) of human genomic DNA either is transcribed into RNA in at least one type of cell or tissue or is involved in some functional aspect of chromatin structure. Much of the noncoding DNA is associated with regulatory functions that affect the expression of the 20,000 protein-coding genes and the many additional genes encoding functional RNAs. Many mutations associated with human genetic diseases lie in this noncoding DNA, where they probably affect regulation of one or more genes. As described in Chapters 16 and 19 through 22, new classes of functional RNAs are being discovered at a rapid pace. Many are encoded by genes whose existence was previously unsuspected. They are now being identified in screens using technologies such as RNA-Seq (see Section 8.2).
About 3% of the human genome consists of highly repetitive sequences referred to as simple-sequence repeats (SSRs). Generally less than 10 bp long, an SSR is sometimes repeated millions of times per cell, distributed in shorter segments of tandem repeats. The most prominent examples of SSR DNA occur in centromeres and telomeres (see Chapter 9). Human telomeres, for example, consist of up to 2,000 contiguous repeats of the sequence GGTTAG. Additional repeats of simple sequences are found throughout the genome. These isolated segments of repeated sequences, often featuring up to a few dozen tandem repeats of a simple sequence, are called short tandem repeats (STRs). Such sequences are the targets of important technologies used in forensic DNA analysis (see Highlight 7-1).
What does all this information tell us about the similarities and differences among individual humans? Within the human population there are millions of single-base variations, called single nucleotide polymorphisms, or SNPs (pronounced “snips”). Each human differs from the next by, on average, 1 in every 1,000 bp. Many of these variations are in the form of SNPs, but a wide range of larger deletions, insertions, and small rearrangements also occur in the human population. From these often subtle genetic differences comes the human variety we are all aware of—such as differences in hair color, stature, eyesight, allergies to medication, foot size, and (to some unknown degree) behavior.
The process of genetic recombination during meiosis tends to mix and match these small genetic variations so that different combinations of genes are inherited (as discussed in Chapter 13). However, groups of SNPs and other genetic differences that are close together on a chromosome are rarely affected by recombination and are usually inherited together; these groupings are known as haplotypes. Haplotypes provide convenient markers for certain human populations and for individuals within populations.
Defining a haplotype requires several steps. First, positions that contain SNPs in the human population are identified in genomic DNA samples from multiple individuals (Figure 8-7a). Each SNP may be separated from the next by many thousands of base pairs. Second, SNPs that are inherited together are compiled into haplotypes (Figure 8-7b). Each haplotype consists of the particular bases found at the various SNP positions in the defined haplotype. Finally, tag SNPs—a subset of the SNPs that define the entire haplotype—are chosen to uniquely identify each haplotype (Figure 8-7c). By sequencing just these tag positions in genomic samples from human populations, researchers can quickly identify which of the haplotypes are present in each individual. Especially stable haplotypes exist in the mitochondrial genome (which never undergoes meiotic recombination) and on the male Y chromosome (only 3% of which is homologous to the X chromosome and thus subject to recombination). As we will see in Section 8.3, haplotypes can be used as markers to trace human migrations.
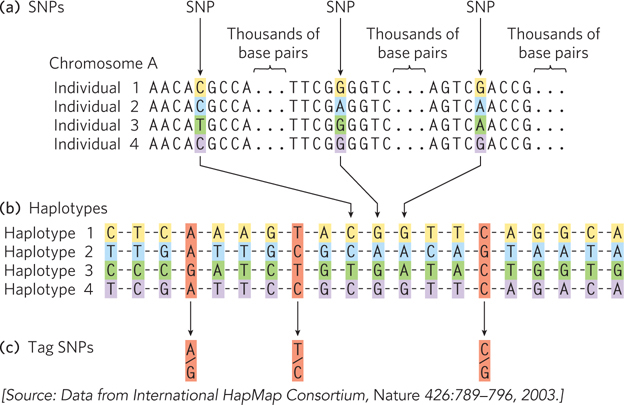
Figure 8-7: Haplotype identification. (a) Positions in the human genome where single nucleotide polymorphisms occur are often identified in genomic samples. The SNPs can be in any part of the genome, whether or not part of a known gene. (b) Groups of these SNPs are compiled into a haplotype. The SNPs will vary in the overall human population, such as in the four fictional individuals shown here, but the SNPs chosen to define a haplotype are often the same in most individuals of a particular population. (c) A few single nucleotide polymorphisms are chosen as haplotype-defining SNPs (tag SNPs), which are then used to simplify the process of identifying an individual’s haplotype (by sequencing 3 loci instead of 20). If the positions shown here are sequenced, an A-T-C haplotype might be characteristic of a population native to a location in northern Europe, whereas a G-T-C haplotype might prevail in a population in Asia. Multiple haplotypes of this kind are used to trace prehistoric human migrations.
Genome Sequencing Informs Us about Our Humanity
A primary purpose of most genome sequencing projects is to identify conserved genetic elements of functional significance, such as conserved exon sequences, regulatory regions, and other genomic features (centromeres, telomeres, etc.). The primary purpose of sequencing the human genome is quite distinct. Here, we are interested in the differences between our genome and those of other organisms. Relying again on the power of evolutionary theory, these differences can reveal the molecular basis of human genetic diseases. They can also help identify genes, gene alterations, and other genomic features that are unique to the human genome and thus likely to contribute to definably human characteristics.
As the genome projects have made clear, the human genome is very closely related to other mammalian genomes over large segments of every chromosome. However, for a genome measured in billions of base pairs, differences of a few percent can add up to millions of genetic distinctions. Searching among these, and utilizing comparative genomics techniques, we can begin to explore the molecular basis of our large brain, language skills, tool-making ability, or bipedalism.
The genome sequences of our closest biological relatives, the chimpanzees and bonobos, offer some important clues and can illustrate the comparative process. Humans and chimpanzees shared a common ancestor about 7 million years ago. Genomic differences between the two species are of two types: base-pair changes (SNPs) and larger genomic rearrangements of many types. SNPs in the protein-coding regions, whether or not they result in an amino acid change, can be used to construct a phylogenetic tree (Figure 8-8a), as described in Section 8.3. Over the course of evolution, segments of chromosomes may become inverted as a result of a segmental duplication, transposition of one copy to another arm of the same chromosome, and recombination between the two segments (Figure 8-8b). Such inversions have occurred in the human lineage on chromosomes 1, 12, 15, 16, and 18. Chromosome fusions can also occur. In the human lineage, two chromosomes found in other primate lineages have been fused to form human chromosome 2 (Figure 8-8c). The human lineage thus has 23 chromosome pairs rather than the 24 pairs typical of simians. Once this fusion appeared in the line leading to humans, it would have represented a major barrier to interbreeding with other primates that lacked it.
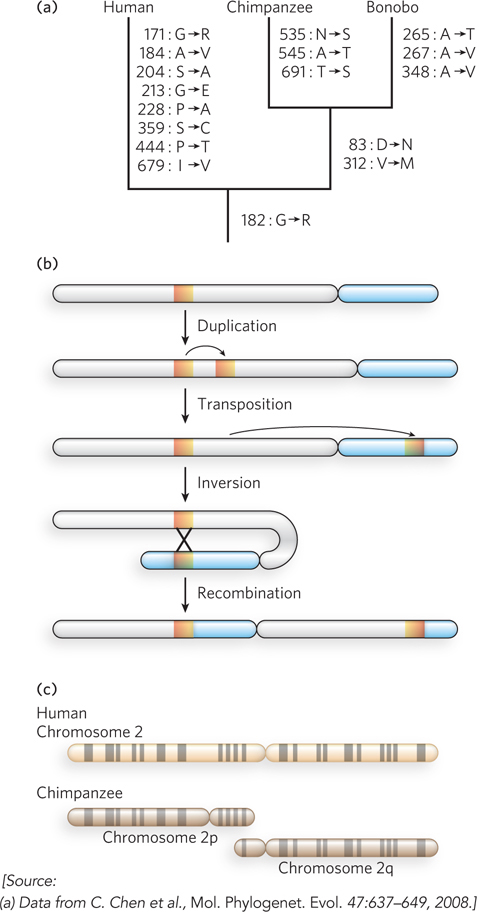
Figure 8-8: Genomic alterations in the human lineage.(a) This evolutionary tree is for the progesterone receptor, which helps regulate many events in reproduction. The gene encoding this protein has undergone more evolutionary alterations than most. Amino acid changes associated uniquely with humans, chimpanzees, and bonobos are listed beside each branch (with the residue number). (b) One of the multistep processes that can lead to the inversion of a chromosome segment. A gene or segment of the chromosome is duplicated, then moved to another chromosomal location by transposition. Recombination of the two segments may result in inversion of the chromosomal DNA between them. (c) The genes on chimpanzee chromosomes 2p and 2q are homologous to those on human chromosome 2, implying that two chromosomes fused at some point in the line leading to humans. Homologous regions can be visualized by bands created in metaphase chromosomes by certain dyes, as shown here.
If we ignore transposons and large chromosomal rearrangements, the published human and chimpanzee genomes differ by only 1.23% at the level of base pairs (compared with the 0.1% variance from one human to another). Some variations are at positions where there is a known polymorphism in either the human or the chimpanzee population, and these are unlikely to reflect a species-defining evolutionary change. When we also ignore these positions, the differences amount to about 1.06%, or about 1 in 100 bp. This might seem a small number, but in large genomes it translates into more than 30 million base-pair changes, some of which affect protein function and gene regulation. Similar calculations show that we are approximately as closely related to bonobos.
The genome rearrangements that help distinguish chimpanzees and humans include 5 million short insertions or deletions involving a few base pairs each, as well as a substantial number of larger insertions, deletions, inversions, or duplications that can involve many thousands of base pairs. When transposon insertions—a major source of genomic variance—are added to the list, the differences between the human and chimpanzee genomes increase. The chimpanzee genome has two classes of retrotransposons that are not present in the human genome (see Chapter 14). Other types of rearrangements, especially segmental duplications, are also common in primate lineages. Duplications of chromosomal segments can lead to changes in the expression of genes contained in these segments. There are about 90 million bp of such differences between humans and chimpanzees, representing another 3% of the genomes. In effect, each species has segments of DNA, constituting 40 to 45 million bp, that are entirely unique to that particular genome, with larger chromosomal insertions, duplications, and other rearrangements affecting more base pairs than do single-nucleotide changes. Thus, the total genomic difference between chimpanzees and humans amounts to about 4% of their genomes.
Sorting out which genomic distinctions are relevant to features that are uniquely human is a daunting task. If the two species share a common ancestor, then, logically, half the changes represent chimpanzee lineage changes and half represent human lineage changes (if one assumes a similar rate of evolution in both lines). When you see a difference, how do you tell which variant was the one present in the common ancestor? One way is to compare both genome sequences with those of more distantly related organisms referred to as outgroups. Consider a locus, X, where there is a difference between the human and chimpanzee genomes (Figure 8-9). The lineage of the orangutan, an outgroup, diverged from that of chimps and humans prior to the chimpanzee/human common ancestor. If the sequence at locus X is identical in orangutan and chimpanzee, this sequence was probably present in the chimpanzee/human ancestor, and the sequence seen in humans is specific to the human lineage. Sequences that are identical in humans and orangutans can be eliminated as candidates for human-specific genomic features. The importance of comparisons with closely related outgroups has given rise to new efforts to sequence the genomes of orangutans, macaques, and many other primate species. And the study of genes and alleles of special significance to humans is being refined as the bonobo genome is further analyzed.
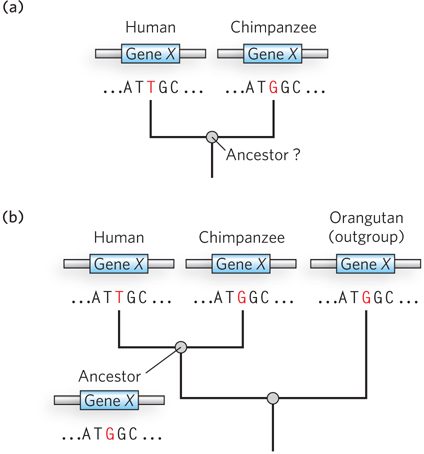
Figure 8-9: Determination of sequence alterations unique to one ancestral line. (a) Sequences from the same hypothetical gene in humans and chimpanzees are compared. The sequence of this gene in their last common ancestor is unknown. (b) The orangutan genome is used as an outgroup. The sequence of the orangutan gene is found to be identical to that of the chimpanzee gene. This means that the mutation causing the difference between humans and chimpanzees almost certainly occurred in the line leading to modern humans, and the common ancestor of humans and chimpanzees (and orangutans) had the variant now found in chimpanzees.
The search for the genetic underpinnings of special human characteristics, such as our enhanced brain function, can benefit from two complementary approaches. The first searches for genomic regions where extreme changes have occurred, such as genes that have been duplicated many times or large genomic segments not present in other primates. The second approach looks at genes known to be involved in relevant human diseases. For brain function, for example, one would examine genes involved in cognition, such as those that contribute to mental disorders when mutated.
Several factors, such as the development of human-specific life history traits (e.g., a greater age of sexual maturity and thus a longer generation time), have led to an approximately 3% slower accumulation of genomic changes in the ancestral line leading to humans than in the line leading to chimpanzees. Evolution has occurred somewhat faster in other primate lines. Observed genetic changes are sometimes concentrated in a particular gene or region. In principle, human-specific traits could reflect changes in protein-coding genes, in regulatory processes, or both. A few classes of protein-coding genes exhibit evidence of accelerated divergence (more amino acid substitutions than normal). These include genes involved in chemosensory perception, immune function, and reproduction. In these cases, rapid evolution is evident in virtually all primate lines, reflecting physiological functions that are critical to all primate species. Another class of genes showing evidence of accelerated evolution is those encoding transcription factors—proteins involved in the expression of other genes (discussed in Chapter 21).
Notably, analyses of the human lineage have not detected an enrichment of genetic changes in protein-coding genes involved in brain development or size. Guided in part by the results obtained for transcription factor genes, the focus of such analyses has gradually shifted to changes in gene expression. In primates, most genes that function uniquely in the brain are even more highly conserved than genes functioning in other tissues. This may reflect some special constraints related to brain biochemistry. However, some differences in gene expression are observed. For example, the gene encoding the enzyme glutamate dehydrogenase, which plays an important role in neurotransmitter synthesis, has an increased copy number due to gene duplication. Analyses of changes in genomic regions related to gene regulation show that genes involved in neural development and nutrition are disproportionately affected. A variety of RNA-coding genes, some with expression concentrated in the brain, also show evidence of accelerated evolution (Figure 8-10). Many of these genes are probably involved in regulating the expression of other genes. The many new classes of RNA that are now being discovered (see Chapter 22) are likely to radically change our perspective on how evolution alters the workings of living systems.
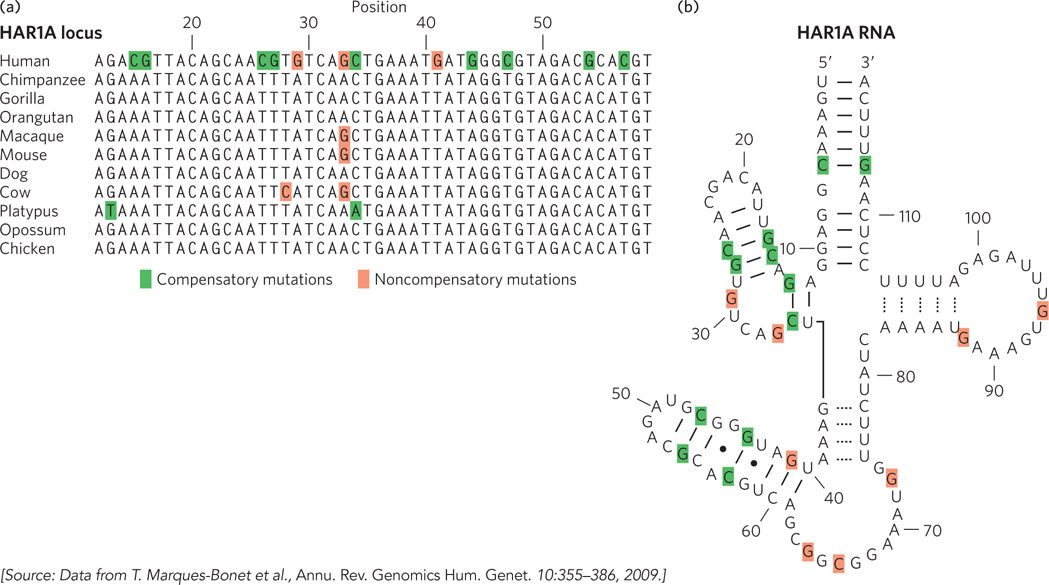
Figure 8-10: Accelerated evolution in some human genes.(a) The HAR1A locus specifies a noncoding RNA that is highly conserved in vertebrates. This RNA functions in the brain during neurodevelopment. In humans, the HAR1A gene exhibits an unusual number of substitutions (highlighted by color shading), providing evidence of accelerated evolution. (b) The secondary structure of the HAR1A RNA has several paired loops. Many of the sequence changes, shaded green here and in (a), are compensatory in the context of this RNA secondary structure: a change on one side of the loop is mirrored by a compensatory change on the other side of the loop, to permit proper base pairing. Noncompensatory changes are shaded red.
Genome Comparisons Help Locate Genes Involved in Disease
One of the motivations for the Human Genome Project was its potential for accelerating the discovery of genes underlying genetic diseases. That promise has been fulfilled: well over 1,600 human genetic diseases have been mapped to particular genes. Some disease-gene hunters caution that so far, the work may have uncovered mostly the relatively easy cases, with many challenges remaining.
The main approach during the past two decades has used a method called linkage analysis. In brief, the gene involved in a disease condition is mapped relative to well-characterized genetic polymorphisms that occur throughout the human genome, using methods firmly rooted in evolutionary biology. The search often begins with one or more large families that include several individuals affected by a particular disease. We illustrate this by describing the search for one gene involved in Alzheimer disease. About 10% of all cases of Alzheimer’s in the United States result from an inherited predisposition. Several different genes have been discovered that, when mutated, can lead to early onset of the disease. One such gene (PS1) encodes the protein presenilin-1, and its discovery made heavy use of linkage analysis. Figure 8-11a shows two of the many family pedigrees that were used to search for this gene in the early 1990s. In studies of this type, DNA samples are collected from both affected and unaffected family members. Researchers first localize the region associated with the disease to a specific chromosome. This effort makes use of a set of genomic locations where common SNPs or other mapped genomic alterations occur in the human population, as identified by the Human Genome Project. Using a panel that includes several well-characterized SNP loci mapped to each chromosome, investigators compare the genotypes of individuals with and without the disease, focusing especially on close family members. By identifying the particular SNPs that are most often inherited with the disease-causing gene, the responsible gene can gradually be localized to a single chromosome. In the case of the PS1 gene, coinheritance was strongest with markers on chromosome 14 (Figure 8-11b).
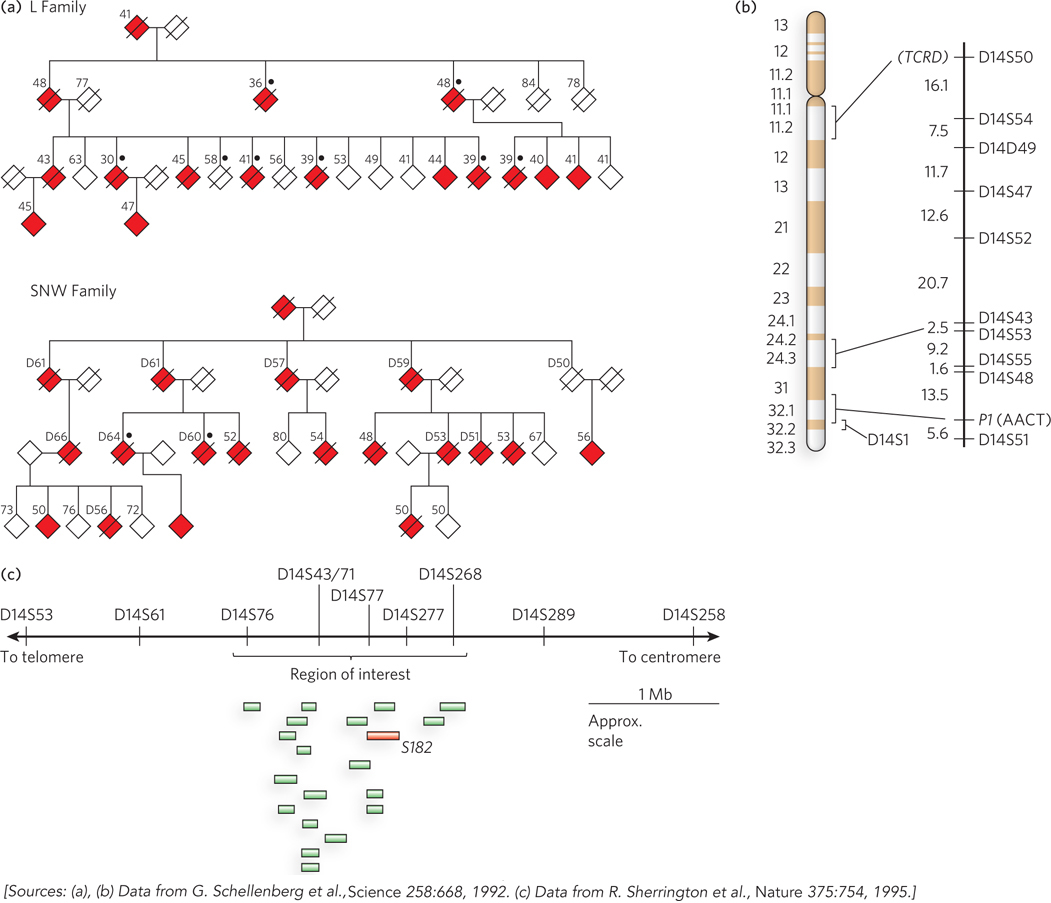
Figure 8-11: Linkage analysis in the discovery of disease genes.(a) These pedigrees for two families affected by early-onset Alzheimer disease are based on the data available at the time of the study. Filled symbols represent affected individuals; slashes indicate deaths. The number above each symbol is the person’s age at onset of symptoms (for affected individuals), or age at the time of the study (for living unaffected individuals), or age at time of death (for deceased unaffected individuals and others marked by “D”). Black dots indicate that an autopsy was done to verify the presence of Alzheimer disease. To protect family privacy, gender is not indicated. (b) Chromosome 14 has marker positions shown at the right, with the genetic distance between them in centimorgans. TCRD (T-cell receptor delta) and PI (AACT, α1-antichymotrypsin) are genes with variations in the human population that were used as markers, along with SNPs, in chromosome mapping. (c) A region of interest containing 19 expressed genes was eventually defined near marker D14S43. The gene labeled S182 (red) encodes presenilin-1. (Mb indicates 106 bases.)
Chromosomes are very large DNA molecules, and localizing the gene to one chromosome is only a small part of the battle. That chromosome contains a mutation that gives rise to the disease, but in every individual human genome, each chromosome contains thousands of SNPs and other changes—representing alterations of all kinds relative to the reference sequence in the human genome database. Simply sequencing the entire chromosome would be unlikely to reveal the SNP or other change associated with the disease. The more detailed localization of a disease-causing gene on a chromosome relies instead on an even more elaborate application of linkage analysis. Statistical methods can correlate the inheritance of additional, more closely spaced polymorphisms with the occurrence of the disease, focusing on a denser panel of polymorphisms known to occur on the chromosome of interest. The more closely a marker is located to a disease gene, the more likely it is to be inherited along with that gene. This process can pinpoint a region of the chromosome that contains the gene. However, the region may still contain a long length of DNA encompassing many genes. In our example of Alzheimer disease, linkage analysis indicated that the disease-causing gene was somewhere near a SNP locus called D14S43 (Figure 8-11c).
The final steps again use the human genome databases. The local region containing the gene is examined and the genes within it are identified. DNA from many individuals, some who have the disease and some who do not, is sequenced over this region. This process, with an increasing number of individuals analyzed, gradually leads to the identification of gene variants consistently present in individuals with the disease state, and not in unaffected individuals. The search can be aided by an understanding of the function of the genes in the target region, because particular metabolic pathways may be more likely than others to produce the disease state. In 1995, the chromosome 14 gene associated with Alzheimer disease was identified as gene S182. The product of this gene was given the name presenilin-1, and the gene itself was subsequently renamed PS1.
Many human genetic diseases are caused by mutations in a single gene or in sequences involved in its regulation, and the defect is inherited in Mendelian patterns (see Chapter 2). Several different mutations in a particular gene, all leading to the same or related genetic condition, may be present in the human population. There are several variants of PS1, for example, all giving rise to a much increased chance of early-onset Alzheimer’s. Another, more extreme example is the several genes encoding different hemoglobins: more than 1,000 known mutational variants are present in the human population. Some of these variants are innocuous; some cause diseases ranging from sickle-cell anemia to thalassemias. The inheritance of particular mutant genes may be concentrated in families or in isolated populations.
More complex are cases where a disease condition is caused by the presence of mutations in two different genes (neither of which, alone, causes the disease), or where a particular condition is enhanced by an otherwise innocuous mutation in another gene. Identifying the genes and mutations responsible for such digenic diseases is exceedingly difficult, and these diseases are sometimes possible to document only within small, isolated, and highly inbred populations.
Modern genome databases are opening up alternative paths to the identification of disease genes. In many cases, we already have biochemical information about the disease. In the case of Alzheimer disease, an accumulation of the amyloid β-protein in limbic and association cortices of the brain is at least partly responsible for the symptoms. Defects in presenilin-1 (and in a related protein, presenilin-2, encoded by a gene on chromosome 1) lead to elevated cortical levels of amyloid β-protein. Focused databases are being developed that catalog such functional information on the protein products of genes, as well as on protein-interaction networks (determined by methods described in Section 8.2), SNP locations, and other data. The result is a streamlined path to the identification of candidate genes for a particular disease. If a researcher knows a little about the kinds of enzymes or other proteins likely to contribute to a disease, these databases can quickly generate a list of genes known to encode proteins with relevant functions, additional uncharacterized genes with orthologous or paralogous relationships to the genes in this list, a list of proteins known to interact with the target proteins or orthologs in other organisms, and a map of gene positions. With the aid of data from some selected family pedigrees, a short list of potentially relevant genes can often be determined rapidly.
These approaches are not limited to human diseases. The same methods can be used to identify the genes involved in diseases—or genes that produce desirable characteristics—in other animals and in plants.
SECTION 8.1 SUMMARY
A genome is one copy of the complete genetic complement of an organism. Thousands of complete genome sequences are now available. The Human Genome Project was undertaken by two competing teams that used different strategies of shotgun sequencing.
The sequencing of a genome is followed by genome annotation, an attempt to summarize the locations and functions of genes and other sequences.
The human genome contains approximately 20,000 genes, fewer than expected. Only 1.1% to 1.4% of the human genome encodes proteins; the remainder is made up of transposons, functional RNA–encoding genes, introns, sequences involved in gene regulation, and tandem repeats of short sequences.
The sequencing of multiple primate genomes is opening windows into human evolution. Genomic alterations that are specific to the human lineage occupy about 4% of our genome, with large genomic rearrangements such as transposon insertions and segmental duplications playing a larger role than single-nucleotide polymorphisms.
Genome sequence databases facilitate the search for genes that specifically contribute to particular traits, and for genes involved in disease.