Mismatch Repair Fixes Misplaced-Nucleotide Replication Errors
DNA polymerase III of E. coli contains a proofreading activity that confers a very low mutation rate of one error in 106 to 108 nucleotides. However, the observed accuracy of replication in E. coli is even higher: one error in 109 to 1010 polymerization events. The additional accuracy comes from an efficient repair process that recognizes and corrects mismatches that escape Pol III.
Mismatched nucleotides incorporated by the replication apparatus are corrected by the mismatch repair (MMR) system, which is conserved in all cell types from bacteria to humans. The mismatches are nearly always corrected to reflect the information in the parent strand. Given that neither strand contains a damaged base, the cell must discriminate between the parental template and the newly synthesized strand, and replace only the nucleotide base in the new strand. Besides mismatches, the E. coli MMR system can also recognize small loops of up to 4 bp of unpaired nucleotides, formed by template slippage during replication or by recombination. Left unrepaired, these small loops of extra DNA result in deletions or insertions. Loops of more than 4 bp are not recognized by the MMR system, and there is no other mechanism to recognize these mistakes. Thus, larger indels are simply not corrected. The MMR system of E. coli includes at least 12 protein components that function in either the strand discrimination reaction or the repair process itself (Table 12-2).
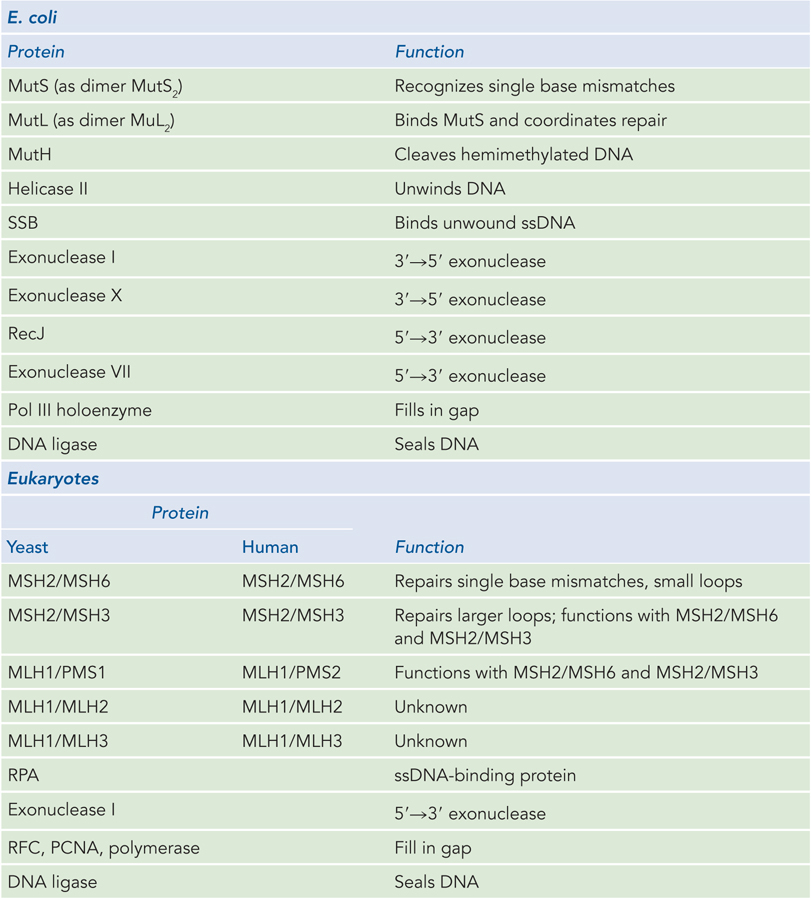
Figure 12-2: Proteins of Mismatch Repair in E. coli and Eukaryotes
The mechanism by which the newly synthesized strand is identified and targeted for correction has not been worked out for most bacteria or eukaryotes, but it the main is well understood for E. coli and some closely related bacterial species. In these bacteria, strand discrimination is based on the action of Dam methylase, the enzyme that methylates DNA at the N6 position of adenine within a 5′-GATC-3′ sequence (see Chapter 11). The GATC sequence on both strands of the parental DNA is methylated, but during replication, the newly synthesized strand is unmethylated for a short period (a few seconds or minutes) immediately after passage of the replication fork. During this interval of hemimethylation, proteins in the MMR complex can identify and distinguish the unmethylated new strand from the methylated parent strand. Replication mismatches in the vicinity of a hemimethylated GATC sequence are then repaired according to the information in the methylated (parental) template strand (Figure 12-17).
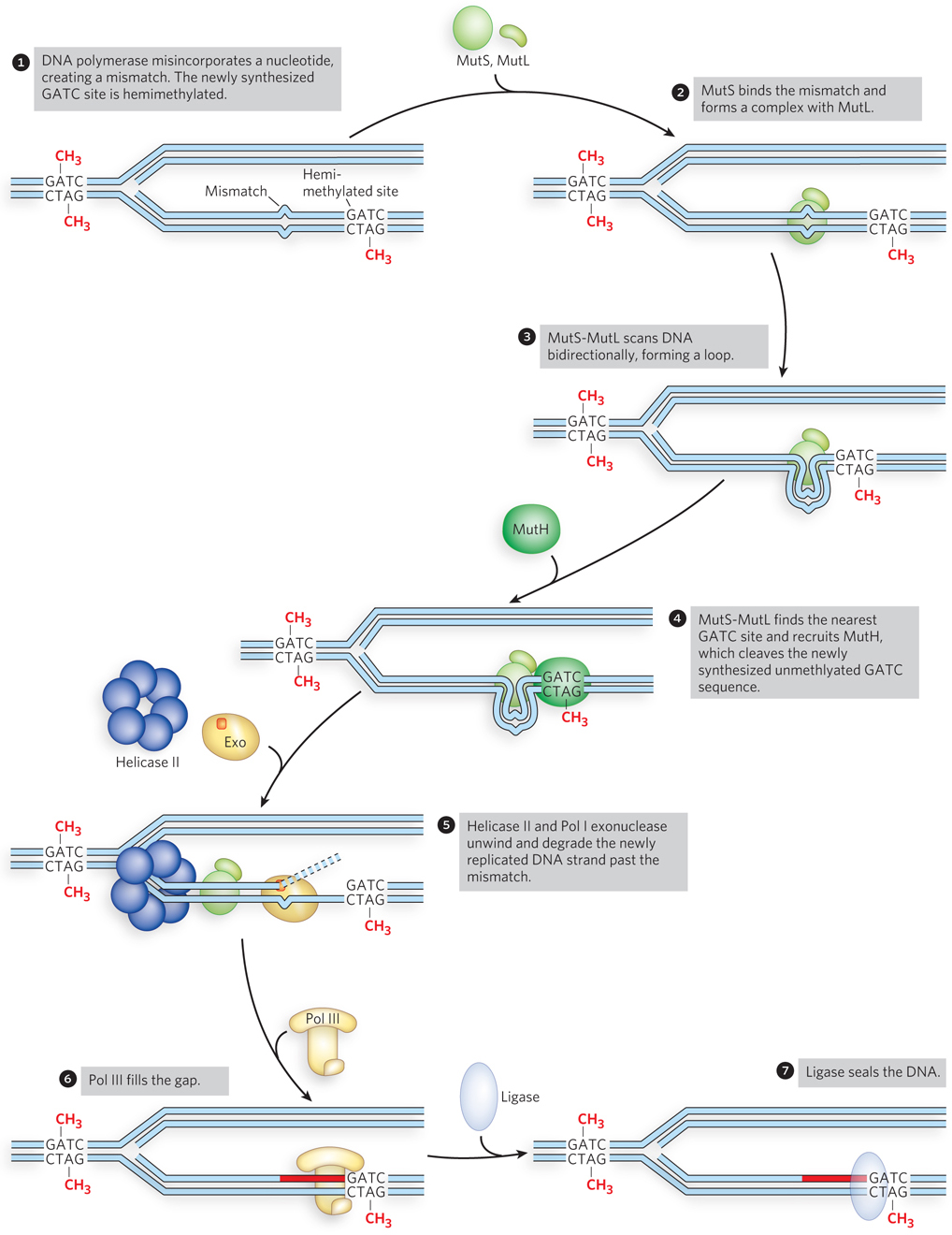
Figure 12-17: Mismatch repair of a nucleotide misincorporated by DNA polymerase. In E. coli, MutS2MutL2 binds a mismatch and scans the DNA for a GATC site. MutH nicks the DNA at the nearest unmethylated GATC site, facilitating repair of the mismatch on the newly synthesized strand by excision, filling in of the gap, and ligation.
Studies of the MMR proteins have outlined the process by which mismatch repair occurs (see the How We Know section at the end of this chapter). The mismatched base pair creates a distortion in DNA that is recognized by the MutS protein. This enables MutS to bind the MutL protein, and the MutS-MutL complex, using ATP, scans bidirectionally along the DNA, forming a DNA loop. The crystal structure of MutS bound to a mismatch shows that MutS forms a homodimer (MutS2) that binds DNA at the dimer interface (Figure 12-18). The MutS dimer also contains a hole large enough to surround the DNA, but whether MutS does encircle DNA during scanning is unknown. On arriving at a hemimethylated GATC site, the complex recruits and activates MutH, a site-specific endonuclease that cleaves unmethylated GATC sites. After strand cleavage, MutS-MutL recruits helicase II (also called UvrD), which unwinds DNA in the direction of the mismatch. During the unwinding, an exonuclease degrades the displaced DNA strand. Different exonucleases are used, depending on whether the enzyme needs to travel in the 5′→3′ or 3′→5′ direction along the DNA (Figure 12-19). Unwinding and DNA degradation stop shortly after the mismatch is excised, leaving a single-strand gap that extends from the mismatch to the original incision at the GATC site. The single-strand gap is coated with single-stranded DNA–binding protein (SSB), filled in by Pol III holoenzyme, and sealed by ligase.
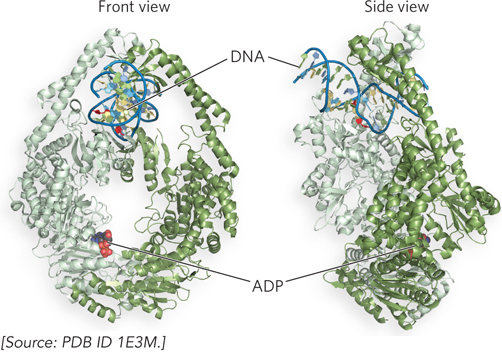
Figure 12-18: The structure of MutS bound to a mismatch in DNA. Front and side views of the MutS homodimer, MutS2 (identical subunits, orange and white), show it enveloping and kinking DNA that contains a mismatched base pair (red). The nucleotide is ADP; ATP is essential for MutS function, but ADP keeps the enzyme from moving on DNA.
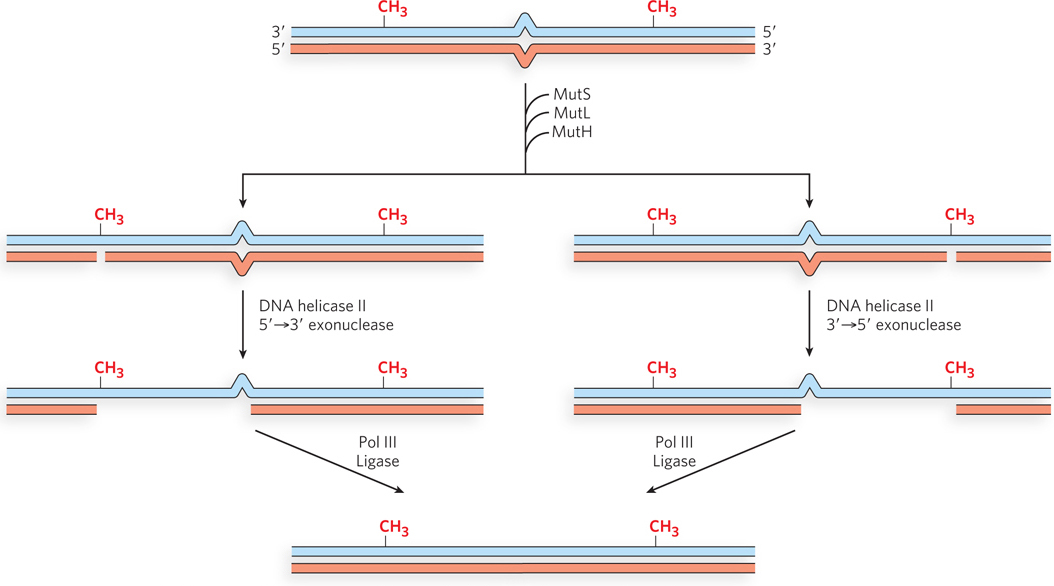
Figure 12-19: Multiple exonucleases involved in methyl-directed MMR. When cleavage at a hemimethylated GATC is on the 3′ side of the mismatch, a 5′→3′ exonuclease is recruited, either RecJ or exonuclease VII. When cleavage is on the 5′ side of the mismatch, a 3′→5′ exonuclease is required, either exonuclease I or exonuclease X.
Mismatch repair is a particularly costly process for E. coli in terms of energy expended. The distance between the mismatch and the GATC cleavage site can be more than 1,000 bp. The degradation and replacement of a strand segment of this length requires an enormous investment in dNTPs to repair a single mismatched base. But this energy consumption is affordable relative to the cost of incurring a mutation. The conservation of such a high-cost repair system in all cells is a good illustration of the importance to cells of maintaining the sequence of their genomic DNA.
Eukaryotic cells have several proteins that are structurally and functionally analogous to bacterial MutS and MutL (see Table 12-2). The MutS homologs work in heterodimers, and each has a specialized function. For instance, in yeast, heterodimers of MSH2 and MSH6 generally bind to single base-pair mismatches and bind less well to slightly longer mispaired loops. The eukaryotic homolog of bacterial MutL is also a heterodimer that binds to the MutS homologs.
Eukaryotic cells lack homologs to bacterial MutH and Dam methylase, and they do not use methylation to distinguish between new and old strands. It is thought that in eukaryotes, strand discrimination relies on the fact that only the newly replicated strand has nicks, and we know that these nicks are frequent on the lagging strand, given the 100 to 200 bp size of Okazaki fragments in eukaryotic cells (see Chapter 11). The nick on a newly synthesized DNA strand can be used as a starting point to excise the DNA strand past the mismatch, and the single-strand gap is then filled in by a polymerase, much as in bacterial mismatch repair; unlike in bacteria, however, the exonucleolytic reaction in eukaryotes does not appear to require the assistance of a helicase. Although this may explain how eukaryotes use nicks to distinguish new from old DNA on the lagging strand, what about the new DNA of the leading strand? The origin of strand nicks in the newly synthesized leading strand is not yet clear, but recent studies show that unlike bacterial MutL, the human MutL homolog has an endonuclease activity that depends on the PCNA clamp, suggesting that strand nicking is coordinated with DNA replication. Because PCNA has distinct “front” and “back” sides, the orientation of a PCNA clamp used during replication may direct the endonuclease to the newly synthesized strand (see Chapter 11).
Mutations in the genes that encode mismatch repair proteins result in the accumulation of mutations throughout the human genome, because misinsertions and short indels can no longer be repaired by the MMR system. Indeed, mutations in MMR genes result in some of the most common inherited cancer-susceptibility syndromes, such as hereditary nonpolyposis colorectal cancer (Highlight 12-1). Approximately 15% of all colon cancers are of this type.
HIGHLIGHT 12-1 MEDICINE: Mismatch Repair and Colon Cancer
Most cancer cells have mutations in genes that regulate cell division (oncogenes and tumor suppressor genes). However, no single mutation is responsible for the progression from a normal cell to a malignant tumor. This progression requires an accumulation of mutations, sometimes over several decades, and is fairly well understood in the case of colon cancer (Figure 1).
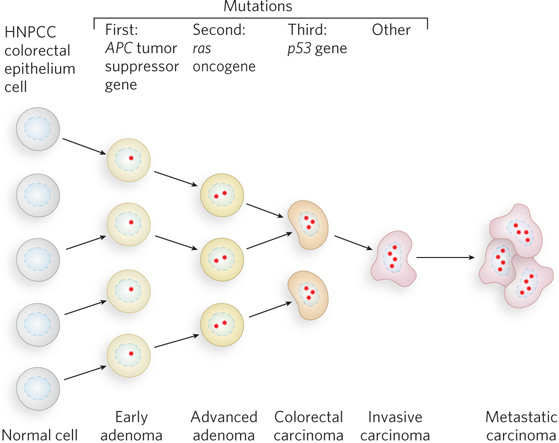
FIGURE 1 The development of colorectal cancer has several recognizable stages, each associated with a mutation. If mismatch repair becomes nonfunctional (through mutation), new mutations accrue quickly.

Richard Kolodner
Discovery of the link between MMR and hereditary nonpolyposis colorectal cancer (HNPCC) was made by the laboratories of Richard Kolodner and Bert Vogelstein, where MMR gene mutations were identified in HNPCC cells. The inherited entity is a loss of function in one allele, usually of the gene encoding MLH1 or MSH2. These genes are essential to mismatch repair (see Table 12-2). Mutation of the second allele leads to the rapid accumulation of multiple new mutations that produce a malignant cell. HNPCC mutant cells have an increased frequency of small insertions and deletions in microsatellite repeats—1 to 6 bp sequences that are repeated 10 to 100 times. This is referred to as microsatellite instability.

Tom Kunkel
The exact number of microsatellite repeats varies from one person to the next, but in one individual, all cells normally contain the identical number of repeats. However, in a person with HNPCC, cells contain different numbers of microsatellite repeats. Independent studies in the laboratories of Tom Kunkel and Paul Modrich showed that extracts of cells displaying microsatellite instability were defective in mismatch repair.
A test of microsatellite length is a simple indication of whether an individual has a mutation in the MMR genes of a tumor (Figure 2). PCR primers are used to amplify specific regions containing microsatellite sequences in the genome. The tumor cell contains some microsatellite DNAs that are longer or shorter than those of a normal cell from the same individual, thus indicating a defective MMR gene. The person was born with two good alleles, but both copies of MLH1 or MSH2 became inactive during his or her lifetime. Microsatellite instability and defects in mismatch repair have now been correlated with several types of cancer other than colon cancer, including ovarian, stomach, cervical, breast, skin, lung, prostate, and bladder cancers.
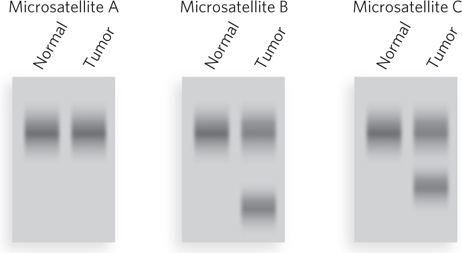
FIGURE 2 PCR primers are designed to amplify genomic DNA of three different microsatellite repeats in normal cells and tumor cells from the same individual. In this illustration of a possible result, two of the three microsatellite repeats that were tested have different sizes in the tumor cells—evidence that the tumor originated from a mutation in an MMR gene.
Direct Repair Corrects a Damaged Nucleotide Base in One Step
Some types of DNA damage that would normally lead to a base substitution or a one-nucleotide deletion are repaired directly, without removing a base or a nucleotide. The best-characterized example of direct repair is the photoreactivation of cyclobutane pyrimidine dimers, first recognized in the late 1940s, before the discovery of DNA structure (see the How We Know section at the end of this chapter). Scientists noticed that bacteria and bacteriophage recovered from UV radiation damage more efficiently when exposed to sunlight. Genetic study of this photoreactivation attributed the repair to a single gene. The gene product is an enzyme referred to as DNA photolyase. Photolyase uses the energy derived from absorbed visible light to reverse the damage of UV light (Figure 12-20). The energy absorbed from visible light by a first chromophore (a light-absorbing chemical) in the enzyme results in electron transfer to a second chromophore, FADH−, to form the free radical FADH·. FADH· donates its electron to the pyrimidine dimer, reversing the cross-links and transferring the electron back to the photolyase to regenerate monomeric pyrimidines and FADH−. Photolyases are present in almost all cells—bacterial, archaeal, and eukaryotic—yet, for some reason, are not present in the cells of placental mammals (including humans).
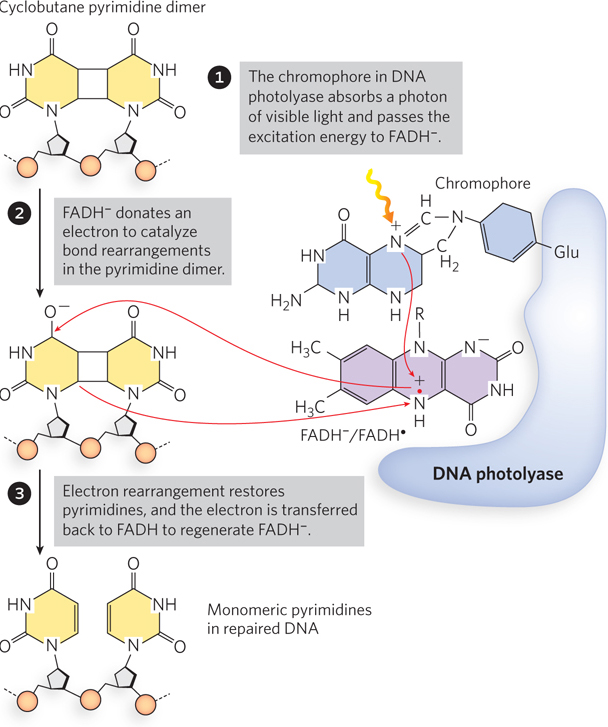
Figure 12-20: Photorepair of a pyrimidine dimer. E. coli photolyase has two chromophores (light-absorbing groups) that work in sequence to use the energy of light to repair a pyrimidine dimer.
More examples of direct repair can be seen in the repair of oxidized nucleotides. The modified base O6-methylguanine is a common and highly mutagenic lesion that results from alkylation (in this case, methylation) of O6 of a G residue. It tends to pair with thymine rather than cytosine during replication, resulting in G≡C to A=T (via O6-meG–T) transition mutations (Figure 12-21a). Direct repair of O6-methylguanine is performed by O6-methylguanine–DNA methyltransferase, an enzyme that catalyzes transfer of the methyl group of O6-methylguanine to one of its own Cys residues (Figure 12-21b). The methyl group transfer leads to irreversible inactivation of the methyltransferase and targets it for degradation (an unusual property for an enzyme). Consumption of an entire protein molecule to correct a single damaged base is another vivid illustration of the priority given to maintaining the integrity of cellular DNA. Direct repair is also used to dealkylate other alkylated nucleotides.
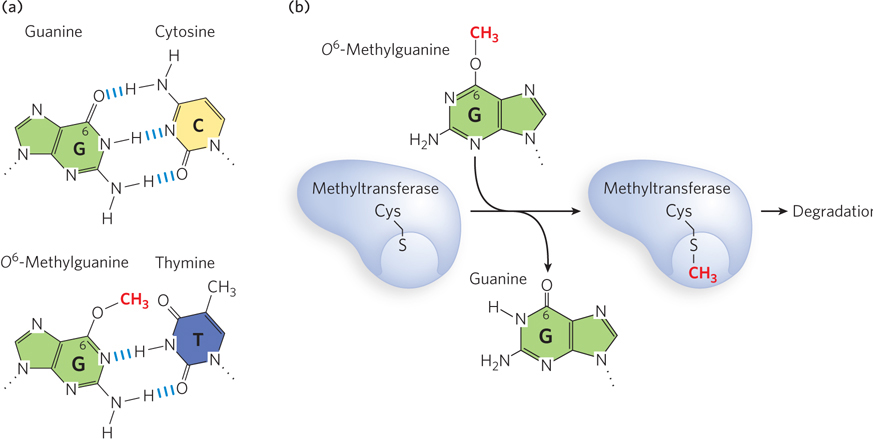
Figure 12-21: Direct repair of methylated nucleotide bases. (a) When the G residue of a normal G≡C base pair (top) is methylated on O6, the resulting O6-methylguanine base-pairs with thymine (bottom) and thus is highly mutagenic. (b) O6-Methylguanine–DNA methyltransferase transfers the methyl group from the O6-methylguanine onto one of its own Cys residues, and the enzyme is thereby targeted for degradation.
Base Excision Repairs Subtle Alterations in Nucleotide Bases
Excision repair is the most prevalent method that cells use to repair damaged DNA. There are two types: base excision and nucleotide excision repair. Base excision repair (BER) functions at the level of a single damaged nucleotide that distorts DNA very little. It is also the main pathway for the repair of single-strand DNA breaks that lack a ligatable junction and therefore require “cleaning” of the 3′ or 5′ terminus for ligation.
In bacterial BER, recognition of the damaged base is performed by a DNA glycosylase, which cleaves the nucleotide base from the pentose by hydrolyzing the N-β-glycosyl bond, leaving an apurinic or apyrimidinic site (AP site). Insertion of the correct nucleotide base does not occur by re-forming the glycosyl bond with a new, correct base. Instead, the single-stranded DNA is cleaved at the abasic site by AP endonuclease, creating a nick with a 3′ hydroxyl and a 5′ deoxyribose phosphate. In E. coli, a segment of DNA is removed by the nick translation activity of Pol I, and DNA ligase seals the remaining nick (Figure 12-22a).
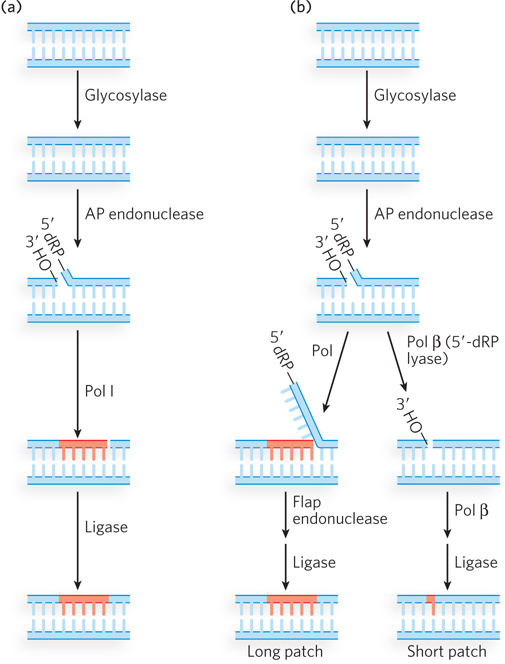
Figure 12-22: Base excision repair. (a) In bacteria, a glycosylase excises a damaged nucleotide base, then an AP endonuclease nicks the backbone at the abasic site. Nick translation by Pol I excises the 5′ deoxyribose phosphate (5′-dRP) and some dNMPs, and synthesizes a new strand. Ligase seals the gap. (b) Eukaryotic BER, after the first two steps (similar to those in bacteria), can take either of two paths. In long patch repair (left), a DNA polymerase extends the DNA strand from the 3′ terminus, displacing the 5′ single-stranded DNA; this is followed by cleavage by a flap endonuclease and ligation. In short patch repair (right), only one nucleotide is inserted (by Pol β) prior to ligation.
Eukaryotic BER proceeds by either of two paths; in each case, the first two steps are the same as in bacteria (Figure 12-22b). One eukaryotic BER mechanism, similar to bacterial BER, is often referred to as long patch repair because up to 10 nucleotides are replaced. Eukaryotic DNA polymerases lack 5′→3′ exonuclease activity, and therefore a special “flap endonuclease” is recruited to remove the displaced 5′ ter minus. The second eukaryotic BER mechanism is used the most; it replaces only the damaged nucleotide base and thus is sometimes referred to as short patch repair. The one-nucleotide fill-in reaction is performed by Pol β, which also removes the 5′ deoxyribose phosphate, leaving a 5′ phosphate for ligation.
Most damaged bases repaired by the BER system remain base-paired in the helix and stack with adjacent bases. This brings up the question of how a damaged base that is buried in the DNA helix can be identified by an enzyme for repair. The crystal structure of a uracil DNA glycosylase (UDG; discussed below) reveals a fascinating recognition process in which the enzyme scans the minor groove of the helix, and damaged-base recognition is performed by kinking the DNA and “flipping” the damaged base completely out of the helix and into the enzyme’s active site (Figure 12-23a). An experiment demonstrating UDG activity is shown in Figure 12-23b. The substrate for this reaction is a synthetic 23-mer duplex in which one strand has an internal dU residue and a 5′-terminal 32P label. The UDG removes the uracil, forming an abasic site in the [32P]DNA strand. Treatment with an AP endonuclease then results in cleavage of the 32P-labeled strand. Analysis in a DNA sequencing gel, which separates the strands, reveals the smaller, cleaved [32P]DNA strand.
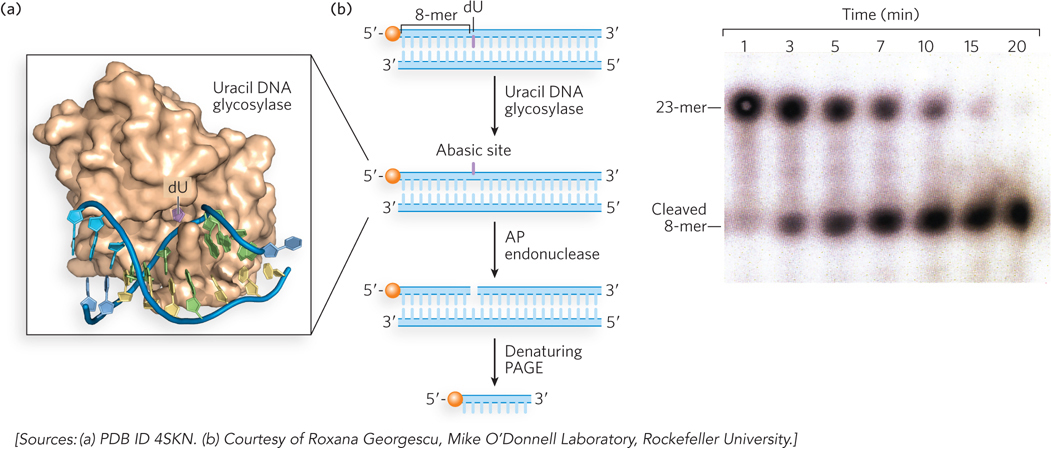
Figure 12-23: Uracil DNA glycosylase. (a) Human uracil DNA glycosylase (brown) bound to its DNA substrate. The uracil (purple) is flipped out of the DNA duplex and fits into the enzyme active site. (b) In this experiment, the DNA duplex, labeled at the 5′ end with 32P (orange balls), is treated with E. coli uracil DNA glycosylase for different time intervals to produce the abasic site, then with AP endonuclease to cleave the phosphodiester backbone. Electrophoresis in a denaturing polyacrylamide gel (PAGE), followed by autoradiography (right), shows results for the various time periods.
All cells contain several different glycosylases that recognize different types of damaged bases (Table 12-3). There are two main types of DNA glycosylases. One type is highly specific to a particular damaged base; the other type recognizes oxidative damage, and the substrate spectrum is more diverse. As discussed earlier, spontaneous deamination of C residues to U residues in DNA is fairly frequent, and chromosomal DNA is in need of constant repair of uracil bases. A uracil DNA glycosylase is found in most cells, and it specifically removes uracil bases from DNA. This glycosylase acts only on DNA; it does not remove uracil from RNA. As may be expected, E. coli strains with mutations in this enzyme have a high rate of G≡C to A=T mutations.
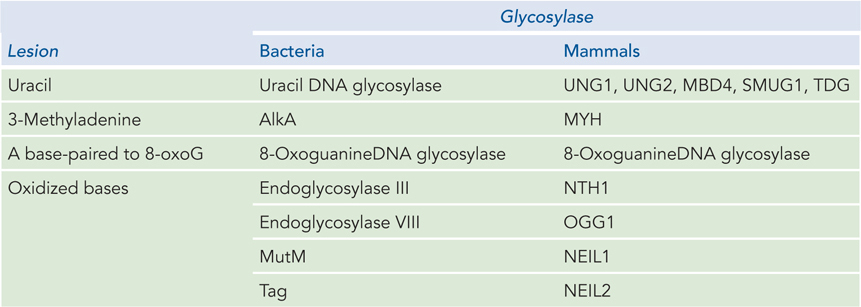
Figure 12-3: DNA Glycosylases in Bacteria and Mammals
Most bacteria have just one UDG, whereas humans and other mammals have several types (see Table 12-3), with different specificities for removing U residues. Specific UDGs remove uracils incorporated during replication, or those formed by deamination of cytosine in double-stranded DNA or single-stranded DNA, or those formed in DNA during transcription. There is also a human DNA glycosylase that removes T residues generated by the deamination of 5-meC. Mismatch repair can also recognize T–G and U–G mismatches, and it corrects them with different levels of efficiency, depending on the sequence context.
A wide variety of damaged bases can be removed by other DNA glycosylases that have evolved to recognize lesions such as formamidopyrimidine and 8-oxoguanine (both arising from purine oxidation), hypoxanthine (arising from adenine deamination), alkylated bases including 3-methyladenine and 7-methylguanine, and even some pyrimidine dimers. The BER pathway can also repair the thousands of abasic sites that arise from spontaneous hydrolysis, as well as breaks in single-stranded DNA that require processing at the 3′ or 5′ terminus before ligation.
Nucleotide Excision Repair Removes Bulky Damaged Bases
Nucleotide excision repair (NER) targets large, bulky lesions and removes DNA on either side of them. In contrast to base excision repair, NER does not require specific recognition of a damaged nucleotide and therefore can remove DNA lesions, even those caused by chemicals that did not exist in the environment until recently. It is the predominant repair pathway for removing pyrimidine dimers, 6-4 photoproducts, and several other bulky base adducts, including benzo[a]pyreneguanine, which is formed on exposure to cigarette smoke (see Figure 12-12b). The nucleolytic activity of the NER system is novel in the sense that two incisions are made in one strand of DNA, excising the lesion and several nucleotides on either side of it; this unique enzymatic activity is called an excinuclease.
In E. coli, the NER pathway makes use of four uvr gene products—UvrA through UvrD—as well as several other factors (Table 12-4; Figure 12-24). First, a UvrA2UvrB complex scans the DNA for damage. When it encounters a bulky damaged base, the strands become separated to form a single-stranded DNA bubble containing the lesion, and UvrA dissociates, leaving UvrB tightly bound to the damaged site. UvrB then recruits the UvrC excinuclease to make incisions in the DNA backbone on the 5′ and 3′ sides of the damaged nucleotide(s). The incisions are precise: the fifth phosphodiester bond on the 3′ side of the lesion and the eighth phosphodiester bond on the 5′ side, generating a fragment of 12 to 13 nucleotides (depending on whether the lesion involves one or two bases) containing the lesion. This oligonucleotide is released through the helicase action of UvrD (also called helicase II). The small gap is then filled by Pol I, and the resulting nick is sealed by ligase.
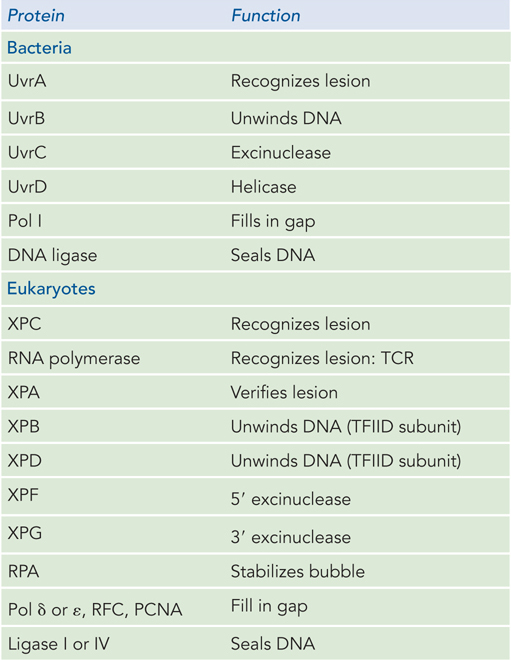
Figure 12-4: Proteins Involved in Nucleotide Excision Repair
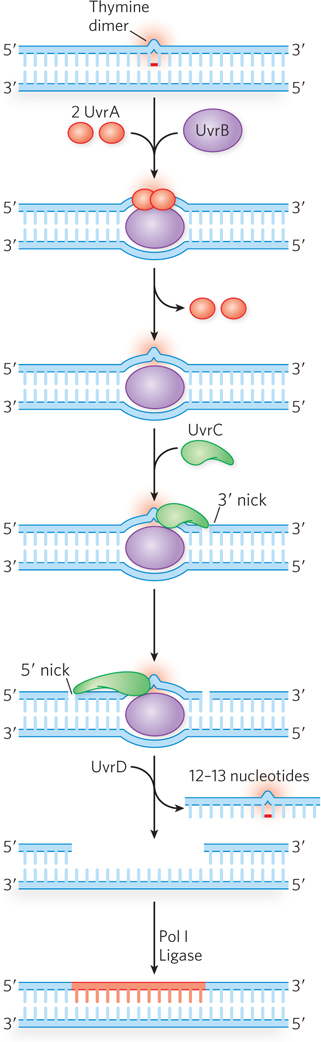
Figure 12-24: Nucleotide excision repair in E. coli. The NER pathway uses several proteins, including UvrA (red), UvrB (purple), and UvrC (green), that recognize the lesion and make incisions on either side, allowing UvrD (helicase II) to displace a section of lesion-containing DNA. The single-strand gap is filled in by Pol I, and the DNA is sealed by ligase. A transcription-coupled repair (TCR) path can also be taken, in which RNA polymerase stalls at the lesion on the coding strand (not shown). After the RNA polymerase is displaced, the reaction proceeds as shown here, using UvrA–D, Pol I, and ligase.
In eukaryotes, NER follows a similar chemical path, although the enzymes are completely different in amino acid sequence and several additional factors are involved (Figure 12-25; see Table 12-4). The main factors were discovered through research on the human genetic disease xeroderma pigmentosum (XP) (Highlight 12-2). Individuals with XP are thousands of times more likely to develop skin cancer from exposure to sunlight. Studies of such individuals have identified at least seven different genes that, when any one of them is defective, can contribute to XP. All of these genes are involved in nucleotide excision repair.
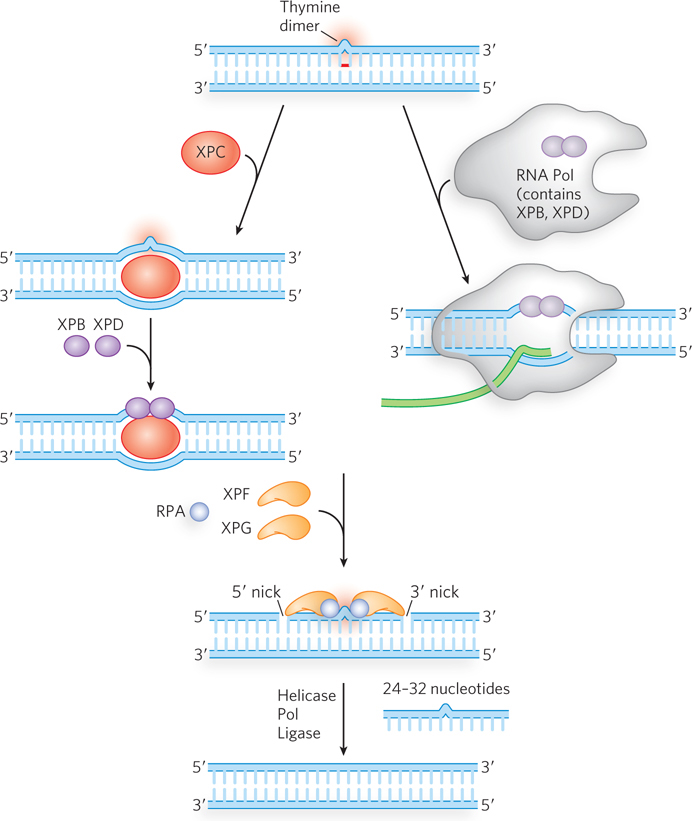
Figure 12-25: Nucleotide excision repair in eukaryotes. NER can be initiated by two slightly different methods in eukaryotes. One pathway (left) is similar to that occurring in bacteria, except that a larger section of lesion-containing DNA is removed. The other pathway (right) is referred to as transcription-coupled repair (TCR), because the lesion is first encountered by RNA polymerase, which then stalls. Some of the NER factors are bound to the RNA polymerase itself.
HIGHLIGHT 12-2 MEDICINE: Nucleotide Excision Repair and Xeroderma Pigmentosum

Robert Painter

James Cleaver
Early studies of UV-damaged E. coli cells showed that their survival was enhanced if the cells were incubated in growth medium before being plated. In genetic studies of this effect, researchers isolated strains with mutations in three different genes, uvrA, uvrB, and uvrC. Further study demonstrated that the repair of UV damage produced small sections of DNA synthesis, which later studies showed were at sites of DNA damage. In Robert Painter’s laboratory at the University of California, San Francisco, a similar process was shown to occur in mammalian cells. James Cleaver, working in Painter’s lab, recognized that the mammalian repair might be related to the repair of UV lesions in bacteria, but he needed similar mutants in mammalian cells to cement this connection. After reading an account of the genetics of human xeroderma pigmentosum (XP), Cleaver realized that the disease might be the sought-after connection to the UV repair pathway in bacteria. He obtained skin biopsies from patients with XP and developed cell lines that could be grown in culture.
Using the XP cell lines and methods for studying nucleotide excision repair in bacteria, Cleaver was able to identify the major protein components of NER in humans. Cell extracts from each patient were used to measure repair. No extract by itself could repair a UV lesion, but when two extracts were mixed together, repair was observed. These extracts, each missing a different protein of the repair pathway, complemented each other. The researchers were eventually able to group types of XP by complementation and identify which protein was missing in each complementation group. Defects in genes encoding any of seven different proteins of NER can result in XP; the proteins are denoted XPA to XPG. Some of these—XPB, XPD, and XPG—also play roles in transcription-coupled repair. Because NER is the sole repair pathway for pyrimidine dimers in humans, people with XP are extremely sensitive to light and readily develop sunlight-induced skin cancers with intense freckling (Figure 1). Most people with XP also have neurological abnormalities, possibly because of an inability to repair lesions caused by the high rate of oxidative metabolism in neurons.
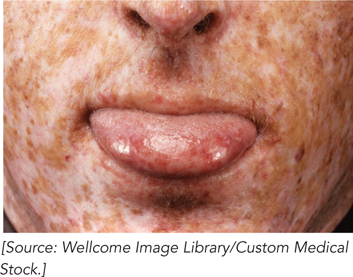
FIGURE 1 This patient exhibits the characteristics of xeroderma pigmentosum.
Various facets of NER in humans await further study. For example, the function of XPE is not yet known. Also, it seems incongruent that bacteria have a second pathway for repair of pyrimidine dimers, making use of DNA photolyase, but humans and other placental mammals do not. However, mammals do have a pathway that bypasses pyrimidine dimers, involving the translesion polymerase, Pol η. This enzyme preferentially inserts two A residues opposite a T–T dimer and does not result in a mutation. Indeed, it is tempting to speculate that the appearance of Pol η replaced the need for photolyase in humans, allowing the photolyase gene to be discarded during evolution.
Genetic studies of XP implicate the genes XPA through XPG in eukaryotic NER. Studies of the proteins encoded by these genes have revealed their roles. The XPC protein initiates the repair process by recognizing the lesion, acting like bacterial UvrA. Then XPB and XPD, which normally act as helicases in RNA transcription, are recruited to the lesion, where they separate the DNA strands to form a single-stranded DNA bubble, acting much like E. coli UvrB. RPA, the eukaryotic equivalent of SSB, then binds to the bubble and positions two nucleases, XPF and XPG, on either side of the lesion. XPG cleaves on the 3′ side and XPF on the 5′ side. The 24- to 32-nucleotide fragment containing the lesion is displaced, and the PCNA clamp recruits a DNA polymerase to fill the gap, which is then sealed by ligase.
In eukaryotes, a process has evolved that targets NER to a damaged template nucleotide that has stalled RNA polymerase. This process, referred to as transcription-coupled repair (TCR), differs from NER only in the way the damaged site is recognized (see Figure 12-25). In TCR, the damage is recognized by the RNA polymerase that is stalled at the lesion. TCR is particularly efficient because it specifically targets repair to actively transcribed DNA that is currently yielding information needed for cell survival, rather than correcting lesions that may lie in vast untranscribed regions of the genome. Bacteria also have a type of transcription-coupled repair. When bacterial RNA polymerase stalls at a lesion, it is displaced by the Mfd helicase, which then recruits the UvrABC proteins for lesion repair.
Recombination Repairs Lesions That Break DNA
Lesions that block the replication fork can lead to cell death if not repaired before the next round of replication. One replisome-blocking lesion in DNA is a double-strand break. The typical route by which polymerase-blocking lesions are repaired is through a high-fidelity homologous recombination pathway (see Chapter 13). Repair by homologous recombination makes use of the sister chromosome to recover the original sequence. The chromosomes are paired, and the lesion can be repaired by using the homologous strand for the correct information. The role of homologous recombination in DNA repair may even have been the main selective force that drove the evolution of recombination enzymes.
Double-strand breaks can also be repaired by nonhomologous recombination, which uses a different set of proteins, conserved from bacteria to humans. Sealing the ends of broken DNA by this process usually incurs deletions or insertions and therefore produces mutations. This pathway, referred to as nonhomologous end joining (NHEJ; see Chapter 13), may be particularly useful when a sister chromosome is unavailable for high-fidelity homologous recombination. Typically, only small deletions or insertions are observed as a result of NHEJ, although large deletions of more than 1 kbp can occur. The sequence at the site of the DNA religation suggests that NHEJ occurs through short, 1 to 6 bp regions of homology.
Specialized Translesion DNA Polymerases Extend DNA Past a Lesion

Wei Yang
Most DNA repair occurs on double-stranded DNA, where the original sequence can be restored using information in the complementary strand. However, sometimes a lesion occurs at a replication fork after the DNA strands have been unwound. In this case, the replicating polymerase stalls at the lesion. One mechanism that has evolved to resolve this potentially lethal situation is a pathway known as translesion synthesis (TLS). Translesion synthesis uses a bypass DNA polymerase, or TLS DNA polymerase, which usually lacks a proofreading 3′→5′ exonuclease and is capable of extending the DNA strand across a bulky template lesion. Structural studies of a TLS polymerase by Wei Yang reveal a wider-than-normal active-site architecture, which may explain how this class of enzymes can misincorporate nucleotides opposite noncoding DNA lesions. Figure 12-26 shows the structure of the complex of an archaeal Pol IV (a TLS polymerase) with DNA containing a benzo[a]pyrene attached to an A residue. The TLS polymerase takes over from the high-fidelity DNA polymerase stalled at the template lesion and extends the DNA strand over the lesion. Because the lesion may be noncoding, lesion bypass by a TLS DNA polymerase often results in a mutation. The first TLS DNA polymerase was discovered in Myron Goodman’s laboratory (see the How We Know section at the end of this chapter).
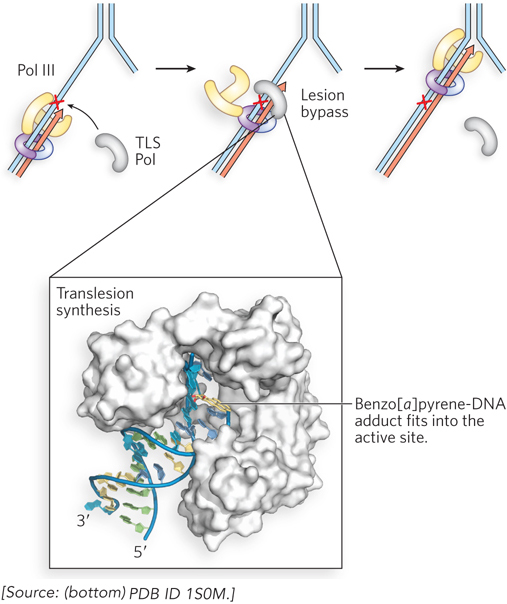
Figure 12-26: Translesion synthesis by TLS DNA polymerases. When the high-fidelity E. coli replicase, Pol III (yellow), encounters a leading-strand lesion (red X), it stalls, and a TLS polymerase (TLS Pol, gray) takes over the β sliding clamp (purple) to extend the leading strand across the lesion. After lesion bypass, Pol III resumes its function with the β clamp. The structure of a TLS DNA polymerase is shown (bottom), the archaeal Dbo4 (white) binding to DNA that contains a benzo[a]pyrene adduct (yellow).
Given the emphasis throughout this chapter on the importance of genomic integrity, the existence of a cellular system that increases the rate of mutation may seem incongruous. However, we can think of this as a desperation strategy. The mutations resulting from translesion synthesis are the biological price a species must pay to overcome an otherwise insurmountable barrier to replication, as it permits a few mutant cells to survive. In E. coli, the main TLS polymerase is Pol V, with components encoded by the umuC and umuD genes. It can extend DNA over the most common lesions, including pyrimidine dimers, 6-4 photoproducts, and abasic sites. All cells contain multiple TLS DNA polymerases (Table 12-5), each suited to bypassing particular types of lesions.
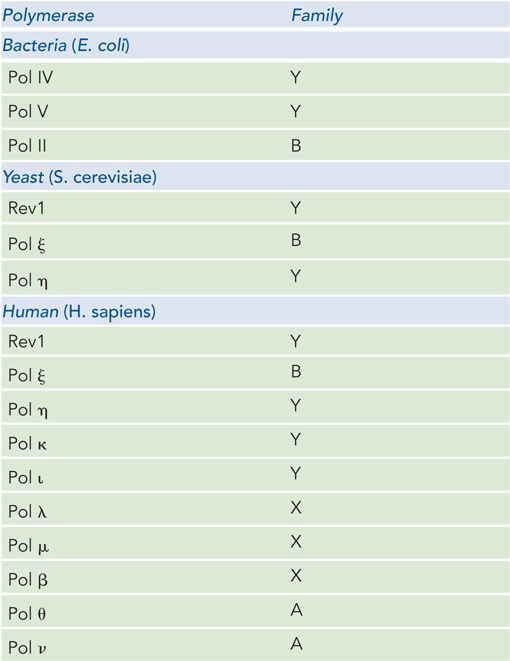
Figure 12-5: Specialized DNA Polymerases Involved in Translesion Synthesis
Humans have at least 10 different TLS polymerases. Pol ξ (xi) is an error-prone TLS polymerase that places nucleotides at random across noncoding lesions. In contrast, Pol η (eta) bypasses pyrimidine dimers in an error-free event, incorporating two A residues opposite the thymine dimer. Pol η goes no farther than the pyrimidine dimer; another DNA polymerase is required to extend the chain to a length that can be used by Pol δ or Pol ε. Pol η has very low fidelity on undamaged DNA, and this may be why another polymerase is needed to extend DNA after Pol η has incorporated A residues opposite a thymine dimer. In other words, Pol η has evolved to be specific and accurate at a thymine dimer, not on normal, unmodified DNA, and therefore any other polymerase will do a better job than Pol η of extending DNA over an undamaged template strand.
Several other low-fidelity DNA polymerases, including Pol β, Pol ι (iota), and Pol λ, have specialized roles in eukaryotic base excision repair. Each of these enzymes has a 5′ deoxyribose phosphate lyase activity in addition to its polymerase activity. After base removal by the glycosylase and backbone cleavage by the AP endonuclease, these low-fidelity polymerases remove the abasic site (a 5′ deoxyribose phosphate) and fill in the gap. The frequency of mutation is minimized by the very short lengths (often just one nucleotide) of DNA synthesized.
This chapter has outlined several of the major types of DNA damage and the DNA repair pathways known to correct, or traverse, the damage. The variety of repair pathways, types of damage they repair, and notable enzymes involved are summarized in Table 12-6.

Figure 12-6: Overview of DNA Repair Processes
SECTION 12.3 SUMMARY
Cells have diverse and robust systems that repair DNA and usually restore it to its original sequence.
The mismatch repair system corrects nucleotide residues misincorporated during replication.
Some types of lesions are repaired directly, such as photoreversal of pyrimidine dimers by photolyase.
The base excision repair pathway corrects relatively small, single-base lesions and uses different DNA glycosylases to recognize particular lesions.
The nucleotide excision repair system repairs bulky lesions by using an excinuclease that makes strand incisions on either side of the lesion. Transcription-coupled repair adapts the nucleotide excision repair system to lesions identified by a stalled RNA polymerase.
Double-strand DNA breaks result in fragmented chromosomes and are usually repaired by homologous recombination, a high-fidelity process. Double-strand breaks can also be processed by error-prone nonhomologous end joining.
Cells contain multiple specialized DNA polymerases that extend DNA across lesions, but translesion synthesis is usually an error-prone process that results in a mutation.
UNANSWERED QUESTIONS
Many mysteries about DNA repair remain to be solved. Basic research in this area currently focuses on the detailed mechanisms of the various pathways. Future studies may well uncover entirely new pathways of DNA repair. We also need better ways of confirming whether particular chemicals are true human carcinogens. Only then can legal controls be put in place as safeguards against these potentially lethal agents in the environment.
Could anticancer drugs overcome the checkpoint controls of the cell cycle and cause the cell to divide before DNA damage has been repaired? Little is known about the detailed workings of how cell cycle checkpoint control is initiated and regulated by DNA damage.
How does DNA repair interweave with other DNA metabolic processes? Repair must be coordinated with other processes of DNA metabolism. For example, enzymes of the recombination, replication, and transcription pathways may encounter lesions, and they need to couple their actions to repair enzymes. Our current understanding of these multiprotein DNA metabolic pathways—transcription, replication, and repair—suggests that they occur in a linear order of independent events. However, it seems likely that the several protein participants in any given pathway are highly coordinated and may even function within some type of superstructure.
How do translesion DNA polymerases coordinate their low-fidelity actions with the high-fidelity polymerase at a replication fork? The primary replicase must have high fidelity, by its very nature. When encountering a template lesion, the replisome must somehow yield to low-fidelity translesion DNA polymerases to carry the DNA across the lesion, otherwise the fork might collapse and the cell could die. How do low-fidelity translesion polymerases take over at a replication fork, yet prevent fork collapse? How does the primary replicase regain access to the replication fork after the translesion polymerase has finished its work?
How does the eukaryotic cell control the timing and activity of all of its many DNA polymerases? The eukaryotic cell contains numerous TLS polymerases. How does the cell know when to use each of these different polymerases? Is the activity of the many DNA polymerases in the cell regulated by posttranslational modification, and if so, how does this affect their function?
HOW WE KNOW: Mismatch Repair in E. coli Requires DNA Methylation
Au, K.G., K. Welsh, and P. Modrich. 1992. Initiation of methyl-directed mismatch repair. J. Biol. Chem. 267:12,142–12,148.
Lahue, R.S., and P. Modrich. 1989. DNA mismatch correction in a defined system. Science 245:160–164.

Paul Modrich
A landmark study by Paul Modrich and his coworkers reconstituted mismatch repair from purified proteins: MutS, MutL, MutH, SSB, helicase II (UvrD), exonuclease I, Pol III holoenzyme, and ligase. A key to success was the insightful design of a circular duplex DNA (with a viral (V) strand and complementary (C) strand) containing a single mismatch and a GATC site about 1 kbp away from the mismatch. The researchers made two different DNAs: one methylated on the V strand and the other methylated on the C strand (Figure 1a).
In two sets of reactions, the two different DNAs were treated with combinations of MutS, MutL, and MutH, followed by agarose gel electrophoresis. One can make the prediction that if the mismatch repair system can distinguish new (unmethylated) from old (parental, methylated) DNA, only the unmethylated strand will be nicked. To distinguish which strand was cleaved, the agarose gel was analyzed with a 32P-labeled DNA probe specific for either the C or V strand (Figure 1b). As a control, the restriction enzyme MboI was used to produce a marker (leftmost lane in Figure 1b), because it cleaves both strands at the GATC site, the same sequence that MutH should nick (on one strand). The result shows that MutH nicks DNA only when MutS and MutL are also present: cleavage is specific to the strand that is unmethylated.
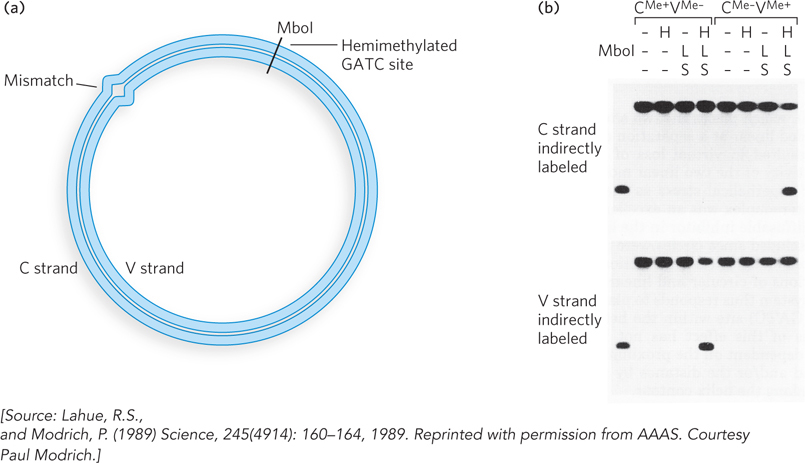
FIGURE 1 MutS and MutL are required to activate MutH, which cleaves the unmethylated strand at a GATC site. (a) The DNA substrate contained a mismatch and a GATC sequence methylated on either the viral (V) strand or the complementary (C) strand. The MboI restriction site (in GATC) is shown. (b) Gel analysis of the reaction products. Proteins added to the reactions in each experiment are indicated at the top of the gel (H, L, and S represent MutH, MutL, and MutS), with MboI fragments as control. DNAs were detected by transfer to a membrane and use of a [32P]DNA oligonucleotide complementary to either the V or the C strand, to produce the autoradiograph.
These studies demonstrate that MutS-MutL activates MutH to cleave the unmethylated strand of a hemimethylated GATC site. Further study using this system showed that the mismatched nucleotide on the nicked strand is corrected in the presence of helicase II, exonuclease I, Pol III holoenzyme, SSB, and ligase. The detailed mechanism of the mismatch repair reaction is described in Section 12.3.
UV Lights Up the Pathway to DNA Damage Repair
Delbecco, R. 1949. Reactivation of ultraviolet-inactivated bacteriophage by visible light. Nature 163:949–950.
Kelner, A. 1949. Effect of visible light on the recovery of Streptomyces griseus conidia from ultraviolet-irradiation injury. Proc. Natl. Acad. Sci. USA 35:73–79.
The first discovery of a DNA repair reaction was that sunlight can repair UV damage. The discovery involved a serendipitous observation by Albert Kelner, an astute scientist who was studying the effects of UV light on the conidia (spores) of the mold Streptomyces griseus, with the hope of finding mutants that produced new varieties of antibiotics. In the course of his studies, Kelner noticed that irradiated cells sometimes had a higher rate of survival (i.e., produced more colonies) than expected. This indicated the presence of a repair pathway.
The variability in survival rate suggested that the repair pathway was inducible. During repeated attempts to identify the conditions that resulted in “induction” of the UV-damage repair pathway, Kelner obtained frustratingly irreproducible results. For example, thinking that temperature influenced the reaction, he incubated cells that received the same dose of UV light at different temperatures. At first there seemed to be an effect, but two preparations treated in the same way gave results that differed more than 100-fold. In a keen observation, Kelner noticed that repair (i.e., increased survival) correlated with culture flasks that were closer to a window, and thus the elusive variable might be sunlight. Controlled experiments confirmed that sunlight was indeed the agent underlying the repair of UV damage. Exposure to visible light yielded more than 105 the number of colonies as the absence of light (Figure 2).
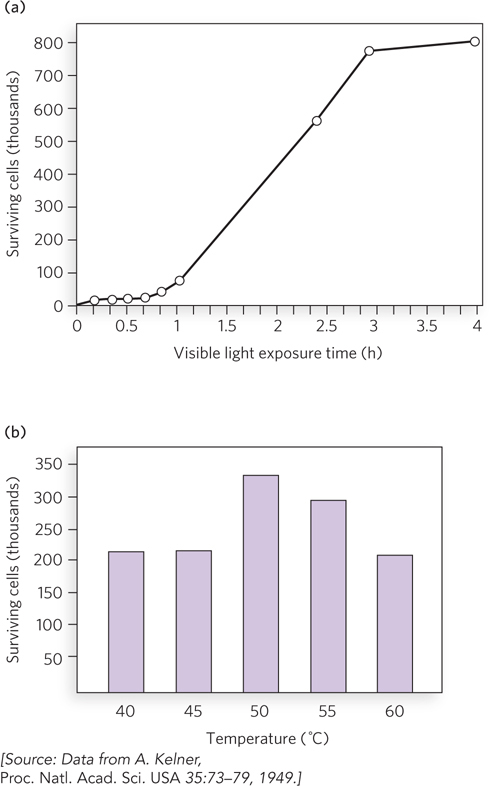
FIGURE 2 Results showing the effect on cell survival of (a) different periods of exposure to visible light and (b) different temperatures of incubation.
Kelner suggested that reversal of UV damage by visible light, or photoreversal, may generalize to other organisms. Having heard of Kelner’s studies, Renato Delbecco, at Indiana University, looked for photoreversal in E. coli using a T phage. UV irradiation of T phage reduced its ability to grow in E. coli, as expected, but its subsequent exposure to visible light had no photoreversal effect on its viability. However, when the UV-irradiated phage were inside E. coli cells and the infected cells were exposed to visible light, Delbecco observed repair (increased phage viability). Hence, he concluded that visible light did not act directly to reverse UV damage, but a cellular factor was required for photorepair.
These studies were performed before scientists knew about DNA structure. We now know that UV light results in pyrimidine dimers and that photolyases directly reverse the pyrimidine cross-links.
Translesion DNA Polymerases Produce DNA Mutations
Rajagopalan, M., C. Lu, R. Woodgate, M. O’Donnell, M.F. Goodman, and H. Echols. 1992. Activity of the purified mutagenesis proteins UmuC, UmuD′, and RecA in replicative bypass of an abasic DNA lesion by DNA polymerase III. Proc. Natl. Acad. Sci. USA 89:10,777–10,781.

Harrison (Hatch) Echols
Bacterial cells contain proteins that produce mutations in response to DNA damage. Four genes are required for this “mutagenic response” in E. coli: recA, lexA, umuC, and umuD. The gene product of umuD is first cleaved by the enzyme RecA coprotease to produce the functional form of UmuD, called UmuD′, which forms a complex with UmuC (UmuC1UmuD′2). Harrison (Hatch) Echols and Myron Goodman intuited that UmuC1UmuD′2 acts during replication, and they developed an in vitro reaction that demonstrates lesion bypass (Figure 3a). They constructed a 5.4 kb, linear, single-stranded DNA substrate with an abasic site near one end, and placed a 5′-32P-labeled primer (P1) just upstream of the abasic site so that they could observe polymerization by PAGE analysis in a denaturing gel. They added RecA, or UmuC, or UmuD′, along with Pol III holoenzyme and SSB, and performed each reaction in duplicate. Bypass of the abasic site is observed only in the presence of UmuC and UmuD′ (Figure 3b; lanes 5 and 6 are duplicate reactions). Later studies showed that UmuC and UmuD′ combine to form a distributive polymerase—one that must dissociate from and rebind to the DNA repeatedly, rather than stick to the DNA constantly as do processive polymerases (such as Pol III). The distributive nature accounts for the many bands in lanes 5 and 6 of the gel analysis. Further study also showed that UmuC is an entirely new class of DNA polymerase, with a sequence unrelated to other known DNA polymerases. The UmuC1UmuD′2 translesion polymerase was renamed Pol V. Soon after these studies, many other translesion DNA polymerases that are homologous to UmuC, designated Y-family polymerases, were identified from cells of all types.
FIGURE 3 Pol V can bypass an abasic site in a DNA substrate. (a) Primer 2 (P2) is added to convert the single-stranded DNA upstream of the lesion to duplex DNA. Primer 1 (P1), labeled with 32P (orange ball), is added to allow observation of extension over the template lesion. (b) PAGE analysis of the reaction products. Only in the presence of RecA, UmuC, and UmuD′ is extension of primer 1 observed (lanes 5 and 6).