18.6 PROTEIN FOLDING, COVALENT MODIFICATION, AND TARGETING
To achieve their biologically active forms, new polypeptides must fold into the correct three-
Protein Folding Sometimes Requires the Assistance of Chaperones
Most proteins fold during translation (after emerging from the ribosome) or immediately after translation, typically beginning with the formation of local secondary structures, including α helices and β sheets. In a cooperative process, these secondary structural elements then interact, often through hydrophobic interactions, to produce the stable three-
After folding into their native conformations, some proteins form intrachain or interchain disulfide bonds, or bridges, between Cys residues. In eukaryotes, disulfide bonds are common in proteins to be exported from cells. The cross-
Covalent Modifications Are Common in Newly Synthesized Proteins
Some newly made proteins, both bacterial and eukaryotic, do not attain their final biologically active conformation until they have been altered by one or more processing reactions known as posttranslational modifications. As we have seen, the first residue inserted in all polypeptides is N-formylmethionine (in bacteria) or methionine (in eukaryotes). The formyl group, the N-
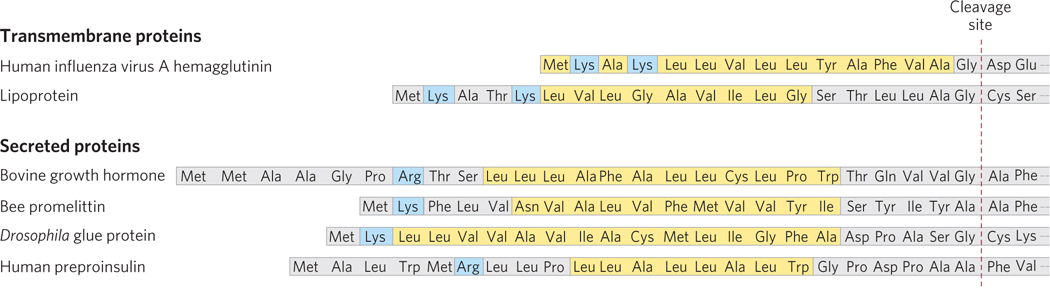
The 15 to 30 residues at the N-
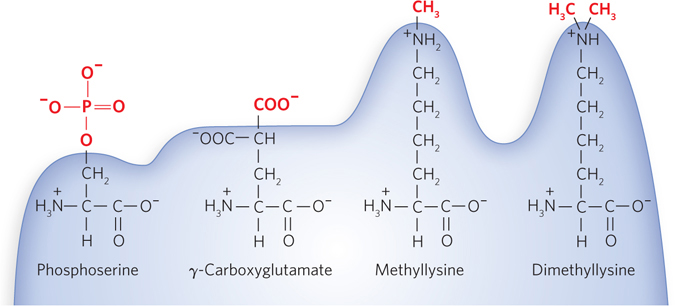
Individual amino acid residues can be modified, either permanently or transiently, with significant effects on functionality, increasing or decreasing the protein’s ability to bind other molecules (Figure 18-34). The hydroxyl groups of certain Ser, Thr, and Tyr residues of some proteins are enzymatically phosphorylated by ATP. Extra carboxyl groups may be added to Glu residues of some proteins, and Lys, Arg, or Glu residues can be methylated.
654
The carbohydrate side chains of glycoproteins are attached covalently during or after synthesis of the polypeptide. In some glycoproteins, the carbohydrate side chain is attached enzymatically to Asn residues (N-linked oligosaccharides), in others to Ser or Thr residues (O-linked oligosaccharides). Many proteins that function extracellularly, as well as the proteoglycans that coat and lubricate mucous membranes, contain oligosaccharide side chains.
Other covalent modifications to proteins include the addition of isoprenyl groups or prosthetic groups and cleavage by proteases. Many bacterial and eukaryotic proteins require covalently bound prosthetic groups for their activity. Finally, many proteins are initially synthesized as large, inactive precursor polypeptides that are proteolytically trimmed to their smaller, active forms.
Proteins Are Targeted to Correct Locations during or after Synthesis

Cells are made up of many structures and compartments, and, in the case of eukaryotic cells, they contain organelles, each with specific functions that require distinct sets of proteins and enzymes. These proteins (with the exception of those produced in mitochondria and chloroplasts) are synthesized on ribosomes in the cytosol or on the endoplasmic reticulum (ER). How are proteins directed to their final cellular destinations? Proteins destined for secretion, integration into the plasma membrane, or inclusion in lysosomes generally share the first few steps of a pathway that begins in the ER. Proteins destined for mitochondria, chloroplasts, or the nucleus use three separate mechanisms. And proteins destined for the cytosol simply remain where they are synthesized.
The most important element in many of these targeting pathways is a short sequence of amino acids called a signal sequence, the function of which was first postulated by Günter Blobel and his colleagues in 1970. The signal sequence directs a protein to its appropriate location in the cell and, for many proteins, is removed during transport or after arrival at its final destination. In proteins directed to mitochondria, chloroplasts, or the ER, the signal sequence is at the N-
Some Chemical Modifications of Eukaryotic Proteins Take Place in the Endoplasmic Reticulum
The best-
655
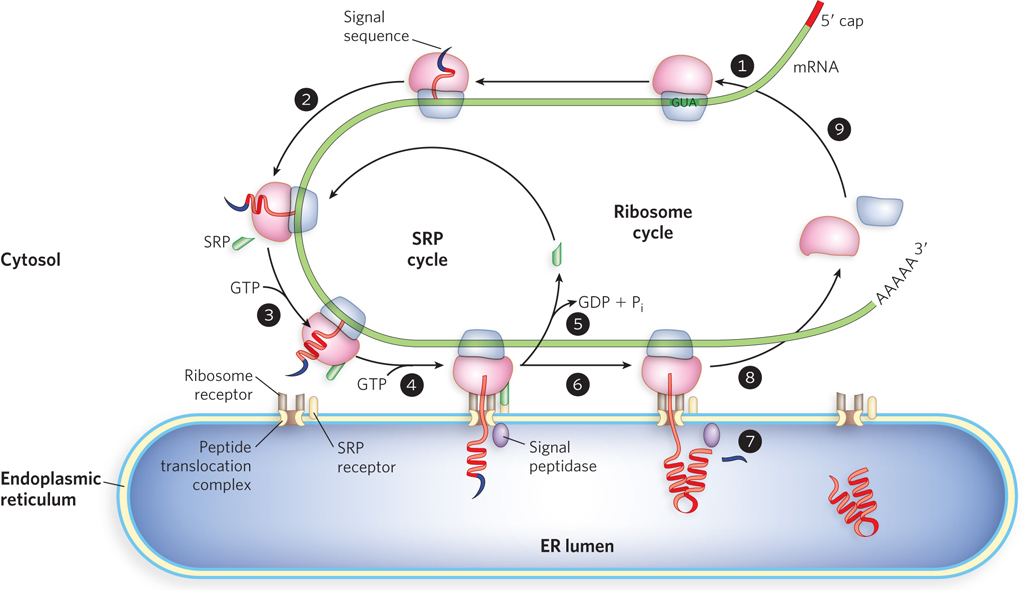
The signal sequence itself helps direct the ribosome to the ER, as shown in Figure 18-36. The targeting pathway begins with initiation of protein synthesis on cytosolic ribosomes (step 1). The signal sequence forms early in the synthesis process: it is at the N-
Glycosylation Plays a Key Role in Eukaryotic Protein Targeting
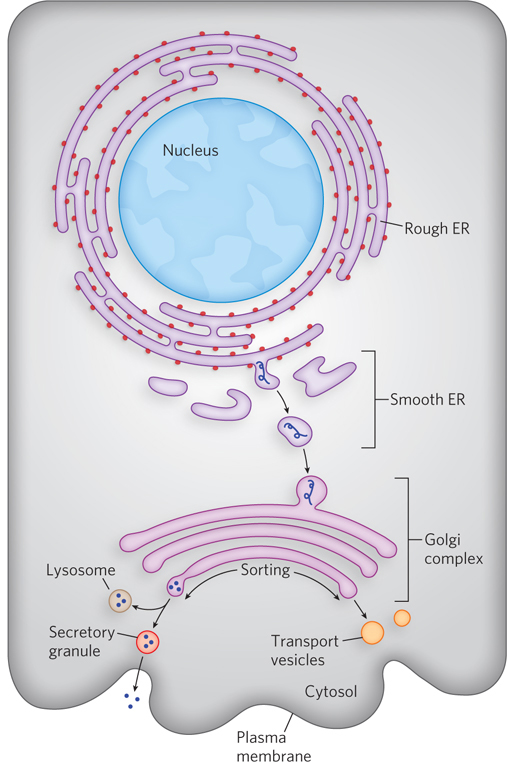
In the ER lumen, newly synthesized proteins are further modified in several ways. Following removal of signal sequences, polypeptides are folded, disulfide bonds are formed, and many proteins are glycosylated. In many glycoproteins, Asn residues are N-linked to a wide variety of oligosaccharides, but the pathways by which they form share common steps. Several antibiotics, including tunicamycin, act by interfering with this process and have aided in elucidating the steps of protein glycosylation. A few proteins are O-glycosylated in the ER, but most O-glycosylation occurs in the Golgi complex (Golgi apparatus) or in the cytosol (for proteins that do not enter the ER).
Once a protein is suitably modified, it can move to its final intracellular destination. Proteins travel from the ER to the Golgi complex in transport vesicles (Figure 18-37). In the Golgi complex, some proteins, as noted above, are O-glycosylated, and some N-linked oligosaccharides are further modified. By mechanisms not yet fully understood, the Golgi complex also sorts proteins and sends them to their final destinations. The processes that segregate proteins targeted for secretion from those targeted for the plasma membrane or lysosomes must distinguish among these proteins on the basis of structural features other than signal sequences, which were removed in the ER lumen.
656
The pathways that target proteins to mitochondria and chloroplasts also rely on N-
Signal Sequences for Nuclear Transport Are Not Removed
Many proteins and nucleic acids move into and out of the nucleus through nuclear pores. RNA molecules synthesized in the nucleus are exported to the cytosol for translation (see Chapter 16). Ribosomal proteins synthesized on cytosolic ribosomes are imported into the nucleus and assembled into 60S and 40S ribosomal subunits in the nucleolus, where the rRNAs are produced. Completed subunits are then exported back to the cytosol. A variety of nuclear proteins are synthesized in the cytosol and imported into the nucleus (e.g., RNA and DNA polymerases, histones, topoisomerases, and proteins that regulate gene expression). All of this traffic is modulated by a complex system of molecular signals and transport proteins.
In multicellular eukaryotes, cell division poses a problem for nuclear proteins. At each cell division, the nuclear envelope breaks down, and after division is completed and the nuclear envelope re-
Nuclear import is mediated by proteins that cycle between the cytosol and the nucleus, including importins α and β and the Ran GTPase (Figure 18-38), in a mechanism like that discussed for nuclear RNA export and import in Chapter 16. A heterodimer of importins α and β functions as a carrier for cargo proteins targeted to the nucleus, with the α subunit binding cargo proteins in the cytosol. The importin-
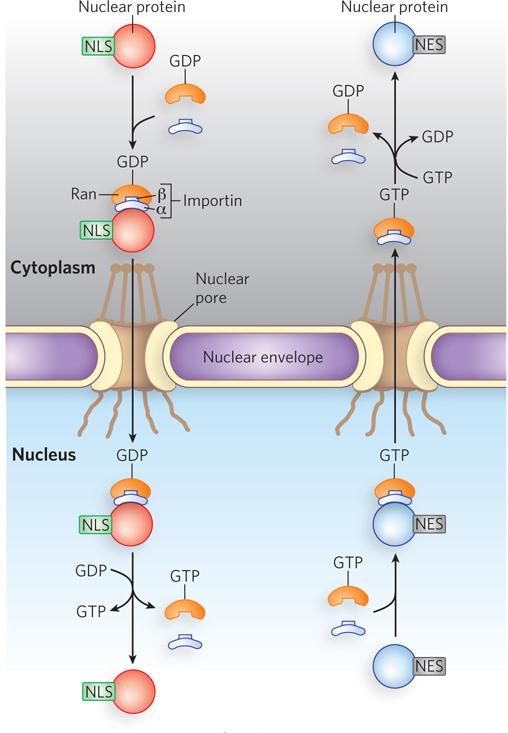
657
Some proteins contain a nuclear export signal (NES), often consisting of four hydrophobic residues that target the protein for export from the cell nucleus to the cytoplasm through the nuclear pore complex. An NES has the opposite effects of an NLS and is recognized by proteins called exportins. The most common spacing of the hydrophobic amino acids of an NES is L-
Bacteria Also Use Signal Sequences for Protein Targeting
Bacteria can target proteins to their inner or outer membranes, to the periplasmic space, or to the extracellular medium. They use signal sequences at the N-
Most proteins exported from E. coli make use of the pathway shown in Figure 18-39. Following translation, the N-
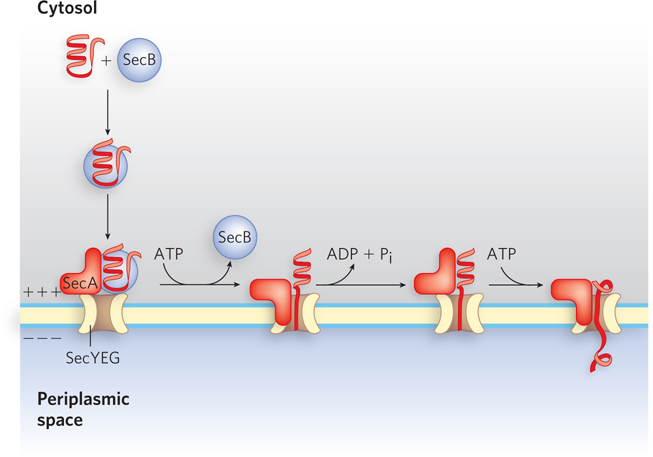
658
SECTION 18.6 SUMMARY
Polypeptides fold into their active, three-
dimensional forms during or immediately after synthesis, often with the help of ATP- dependent chaperone proteins. Many proteins are further processed by posttranslational modification reactions that add functional groups, such as phosphates or sugars. During or immediately following synthesis, many proteins are directed to specific cellular locations. One targeting mechanism involves a peptide signal sequence, generally at the N-
terminus of a newly synthesized protein. In eukaryotic cells, one class of signal sequences is recognized by the signal recognition particle, which binds the signal sequence as soon as it appears on the ribosome and transfers the entire ribosome and incomplete polypeptide to the ER. The peptides are moved into the ER lumen, where they may be modified and moved to the Golgi complex, then sorted and sent to lysosomes, the plasma membrane, or transport vesicles.
Proteins targeted to mitochondria and chloroplasts, and those destined for export in bacterial cells, also make use of an N-
terminal signal sequence. Specific enzymes remove the signal once the protein reaches its destination. Nuclear localization signals are not removed, because nuclear proteins must be relocalized to the nucleus each time the cell divides. Protein import requires a nuclear localization signal (NLS), importins, the Ran GTPase, and GTP. Protein export also requires a nuclear export signal (NES) and exportins.
Bacterial proteins may be targeted to the plasma membrane by a signal sequence.
UNANSWERED QUESTIONS
Although many details of bacterial protein synthesis are known in exquisite detail, researchers have yet to determine how eukaryotic translation is initiated and regulated, how translation assists in protein folding, and how ribosomes work together within polysomes.
How is the initiation of eukaryotic translation regulated? Translation initiation is much more complex in eukaryotic cells than in bacterial cells. It is critical to understand how the eukaryotic system works, because much protein synthesis regulation occurs at the level of initiation. Molecular structures of the eukaryotic ribosome, coupled with more detailed biochemical studies, will help reveal the details of this process.
How is translation rate coupled to protein folding? The physics and kinetics of translation clearly affect how proteins fold. For example, many mRNAs include rare codons for which there are few available matching tRNAs, and these serve to stall ribosomes and hence provide time for proteins to fold. Understanding how this works is important for determining how proteins attain their correct structure in cells. Forces produced by ribosomes as they traverse an mRNA may also be important for melting RNA structures that could otherwise impede translation.
How do the crowded conditions inside cells influence translation rates and accuracy? Most of the experiments that have probed translation mechanisms have been performed using purified ribosomes under relatively dilute conditions (∼1 μm). In rapidly growing cells, however, ribosomes may be present at up to 100 times this concentration. Computer simulations that model the process of translation in the presence of various cellular factors may help determine the impact of molecular crowding on the rate of protein synthesis.
659
HOW WE KNOW: The Ribosome Is a Ribozyme
Noller, H.F., and J.B. Chaires. 1972. Functional modification of 16S ribosomal RNA by kethoxal. Proc. Natl. Acad. Sci. USA 69:3115–
Noller H.F., V. Hoffarth, and L. Zimniak. 1992. Unusual resistance of peptidyl transferase to protein extraction procedures. Science 256:1416–
A series of experiments conducted in the early 1970s by Harry Noller provided the first evidence that ribosomal RNA, rather than ribosomal protein, is responsible for catalyzing peptide bond formation during protein synthesis. Using chemicals that react with side chains in proteins or nucleotides, Noller discovered that the 30S ribosomal subunit of bacteria could be inactivated by a reagent called kethoxal, which primarily attacks guanosine nucleotides in RNA.
Bacterial ribosomes were purified and separated into their two subunits. After addition of kethoxal to the 30S subunits, these were mixed with unmodified 50S subunits and the ribosomes tested for the ability to stimulate in vitro protein synthesis, using tRNA and a poly(U) mRNA template. In contrast to samples with untreated 30S subunits, the kethoxal-
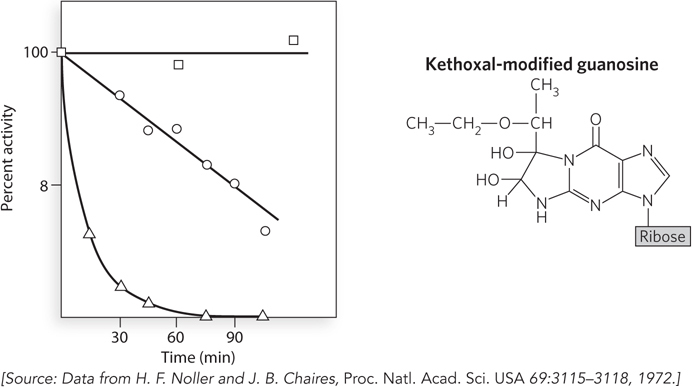
In 1992, Noller and his colleagues published a study showing that virtually all the r-
660
HOW WE KNOW: Ribosomes Check the Accuracy of Codon-
Chapeville, F., F. Lipmann, G. Von Ehrenstein, B. Weisblum, W.J. Ray Jr., and S. Benzer. 1962. On the role of soluble ribonucleic acid in coding for amino acids. Proc. Natl. Acad. Sci. USA 48:1086–
Zaher, H.S., and R. Green. 2009. Quality control by the ribosome following peptide bond formation. Nature 457:161–

Translation relies on aminoacyl-
This experiment, performed in 1962, used 14C labeling to track how amino acids attached to particular tRNAs were incorporated into polypeptides. For example, correctly charged [14C]Cys-
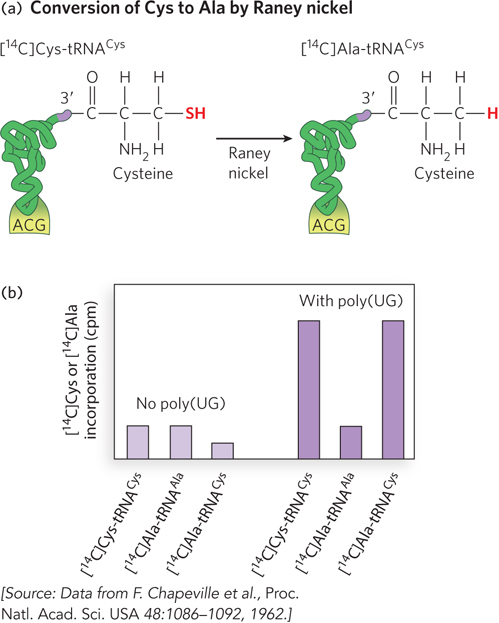
For many years, protein synthesis was thought to rely on the combined accuracy of tRNA aminoacylation and aminoacyl-
More recently, an additional mechanism occurring after peptidyl transfer was found to contribute to the accuracy of protein synthesis. Using a well-
661