Concept 12.2: Prokaryotic Genomes Are Small, Compact, and Diverse
When DNA sequencing became possible in the late 1970s, the first life forms to be sequenced were the simplest viruses. The sequences quickly provided new information about how viruses infect their hosts and reproduce. The next genomes to be fully sequenced were those of prokaryotes. We now have genome sequences for many microorganisms, to the great benefit of microbiology and medicine.
Prokaryotic genomes are compact
In 1995 a team led by Craig Venter and Hamilton Smith published the first complete genomic sequence of a free-living cellular organism, the bacterium Haemophilus influenzae. Many more prokaryotic sequences have followed. These sequences reveal not only how prokaryotic genes are organized to perform different cellular functions, but also how certain specialized functions of particular organisms are carried out.
There are several notable features of bacterial and archaeal genomes:
- They are relatively small. Prokaryotic genomes range in size from about 160,000 to 12 million bp and are usually organized into a single circular chromosome.
- They are compact. Typically, more than 85 percent of the DNA consists of protein-coding regions or RNA genes, with only short sequences between genes.
- Their genes usually do not contain introns. An exception is the rRNA and tRNA genes of archaea, which are frequently interrupted by introns.
- In addition to the main chromosome, they often carry smaller, circular DNA molecules called plasmids, which may be transferred between cells (see Concept 8.4).
Beyond these broad similarities, there is great diversity among these single-celled organisms, reflecting the huge variety of the environments where they are found.
Let’s look in more detail at a few prokaryotic genomes in terms of functional and comparative genomics.
Functional Genomics
As mentioned above, functional genomics is a biological discipline that assigns functions to DNA sequences. This field is less than 20 years old but is now a major occupation of biologists. You can see the various functions encoded by the genomes of three prokaryotes (in this case, all bacteria) in TABLE 12.1.
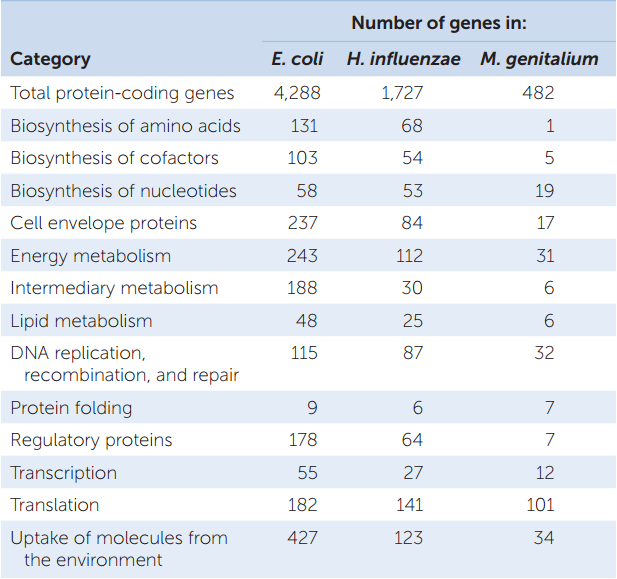
H. influenzae lives in the upper respiratory tracts of humans and can cause ear infections and (more seriously) meningitis. Its single circular chromosome has 1,830,138 bp. In addition to its origin of replication and the RNA genes, this bacterial chromosome has 1,727 open reading frames.
When this sequence was first announced, only 1,007 (58 percent) of the open reading frames encoded proteins with known functions. Since then scientists have identified the role of almost every protein encoded by the H. influenzae genome. All of the major biochemical pathways and molecular functions are represented.
Comparative Genomics
Soon after the sequence of H. influenzae was announced, the smaller Mycoplasma genitalium (580,073 bp) and the larger E. coli (4,639,221 bp) genomic sequences were completed. Thus began the new era of comparative genomics. Scientists can identify genes that are present in one bacterium and missing in another, allowing them to relate these genes to bacterial function.
For example, E. coli has more genes than H. influenzae in each of the functional groups listed in Table 12.1. This suggests that there may be more biochemical pathways in E. coli than in H. influenzae. M. genitalium lacks most of the enzymes needed to synthesize amino acids, which E. coli and H. influenzae both possess (see Table 12.1). This finding reveals that M. genitalium must obtain its amino acids from its environment (usually the human urogenital tract). Furthermore, E. coli has dozens of genes for regulatory proteins that encode transcriptional activators or repressors; M. genitalium has only seven such genes. This suggests that the biochemical flexibility of M. genitalium is limited by its relative lack of control over gene expression.
241
Metagenomics reveals the diversity of viruses and prokaryotic organisms
If you take a microbiology laboratory course, you will learn how to identify various prokaryotes on the basis of their growth in lab cultures. Microorganisms can be identified by their nutritional requirements or the conditions under which they will grow (such as aerobic versus anaerobic). For example, staphylococci are a group of bacteria that inhabit skin and nasal passages. Unlike many bacteria, staphylococci can use the sugar alcohol mannitol as an energy source and thus can grow on a special medium containing mannitol. Often a dye is included in the medium, which changes color if the bacteria are pathogenic (disease-causing). Such culture methods have been the mainstay of microbial identification for more than a century and are still useful and important. However, scientists can now use PCR and modern DNA analysis techniques to analyze microbes without culturing them in the laboratory.
In 1985 Norman Pace, then at Indiana University, came up with the idea of isolating DNA directly from environmental samples. He used PCR to amplify specific sequences from the samples to determine whether particular microbes were present. The PCR products were sequenced to explore their diversity. The term metagenomics was coined to describe this approach of analyzing genes without isolating the intact organism. It is now possible to do DNA sequencing with samples from almost any environment. The sequences can be used to detect the presence of previously unidentified organisms as well as known microbes (FIGURE 12.6). For example:
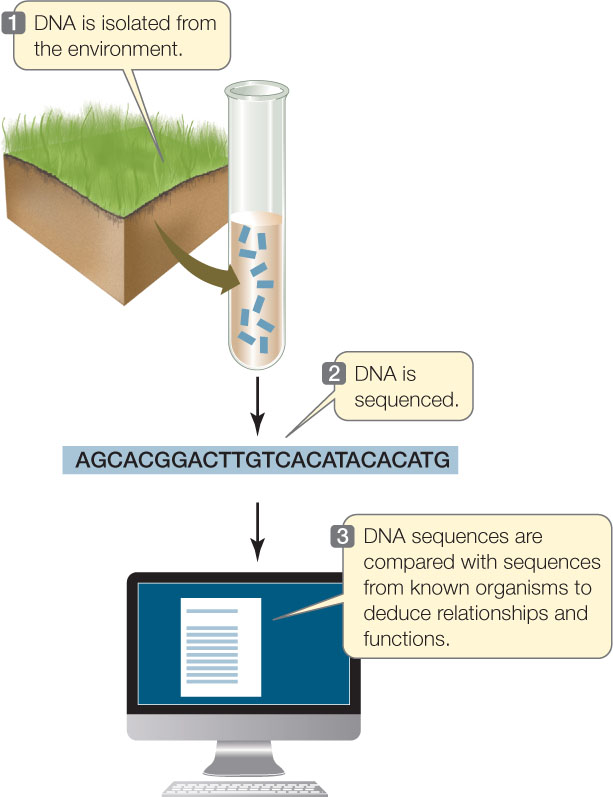
- Sequencing of DNA from 200 liters of seawater indicated that it contained 5,000 different viruses and 2,000 different bacteria, many of which had not been described previously.
- Samples from the intestines of chickens and turkeys from different flocks have led to the identification of viral causes of serious diseases in these domesticated birds.
- Water runoff from a mine contaminated with toxic chemicals contained many new species of prokaryotes thriving in this apparently inhospitable environment. Some of these organisms exhibited metabolic pathways that were previously unknown to biologists. These organisms and their capabilities may be useful in cleaning up pollutants from the water.
- Gut samples from 124 Europeans revealed that each person harbored at least 160 species of bacteria (constituting their gut microflora or microbiome). Many of these species were found in all of the individuals, but the presence of other bacteria varied from person to person. Such variations in gut microflora may be associated with obesity or bowel diseases.
LINK
For more on the complex microbial ecosystem inside the human gut, see Figures 19.20 and 41.1
These and other discoveries are truly extraordinary and potentially very important. It is estimated that 90 percent of the microbial world has been invisible to biologists, in part because the cells could not be grown in the laboratory. These organisms are only now being revealed by metagenomics. Entirely new ecosystems of bacteria and viruses are being discovered in which, for example, one species produces a molecule that another metabolizes. It is hard to overemphasize the importance of such an increase in our knowledge of the hidden world of microbes. This new knowledge underscores the remarkable diversity among prokaryotic organisms, and will further our understanding of natural ecological processes. Furthermore, it has the potential to help us find better ways to manage environmental catastrophes such as oil spills, or to remove toxic heavy metals from soil and water.
Some sequences of DNA can move about the genome
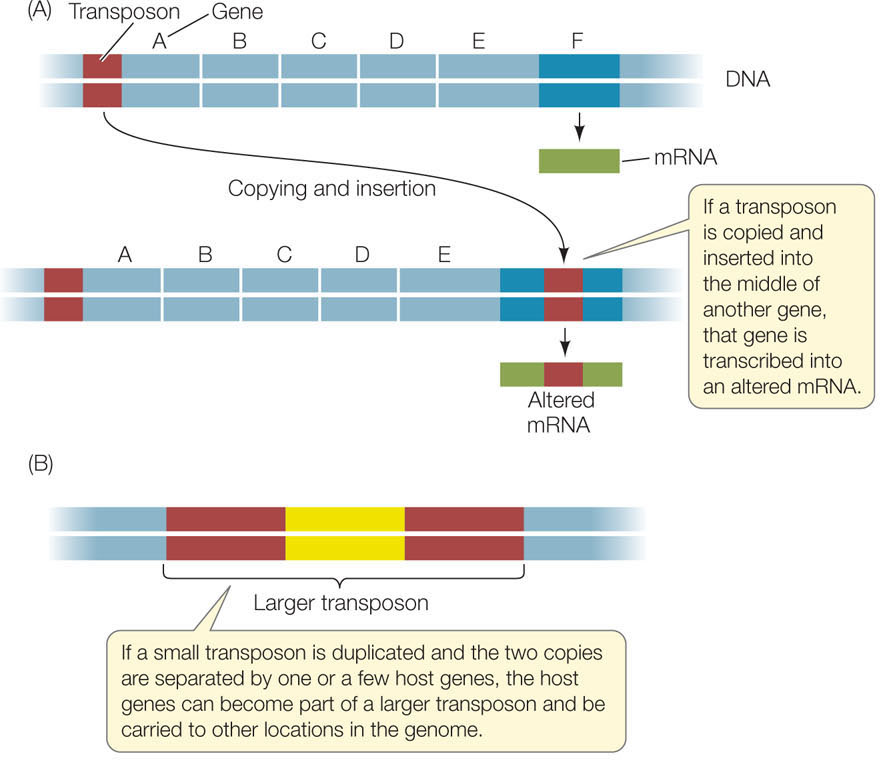
Genome sequencing allowed scientists to study more broadly a class of DNA sequences that had been discovered by geneticists decades earlier. Segments of DNA called transposons (or transposable elements) can move from place to place in the genome and can even move from one piece of DNA (such as a chromosome) to another (such as a plasmid) in the same cell. A transposon might be at one location in the genome of one E. coli cell, and at a different location in another cell. The insertion of this movable DNA sequence from elsewhere in the genome into the middle of a protein-coding gene disrupts that gene (FIGURE 12.7A). Any mRNA expressed from the disrupted gene will have the extra sequence, and the protein will be abnormal. Consequently transposons can produce significant phenotypic effects by inactivating genes.
242
Transposons are often short sequences of 1,000–2,000 bp and are found at many sites in prokaryotic genomes. The mechanisms that allow them to move vary. For example, the transposon may be replicated, and then the copy inserted into another site in the genome. Or the transposon might splice out of one location and move to another location.
If a transposon becomes duplicated, with two copies separated by one or a few genes, the result may be a single larger transposon (up to about 5,000 bp). In this case, the additional genes can be carried to different locations in the genome (FIGURE 12.7B). Some of these transposons carry genes for antibiotic resistance. We will discuss transposons again in Concept 12.3.
Will defining the genes required for cellular life lead to artificial life?
When the genomes of prokaryotes and eukaryotes are compared, a striking conclusion arises: certain genes are present in all organisms (universal genes). There are also some (nearly) universal gene segments that are present in many genes in many organisms. One example is a sequence encoding an ATP binding site, which is a domain found in many proteins. These findings suggest that there is some ancient, minimal set of DNA sequences common to all cells. One way to identify these sequences is to look for them in computer analyses of sequenced genomes.
Another way to define the minimal genome is to take an organism with a simple genome and deliberately mutate one gene at a time to see what happens. M. genitalium has one of the smallest known genomes, with only 482 protein-coding genes. Even so, some of its genes are dispensable under some circumstances. For example, it has genes for metabolizing both glucose and fructose, but it can survive in the laboratory on a medium containing only one of these sugars. Under such circumstances, the bacterium doesn’t need the genes for metabolizing the other sugar.
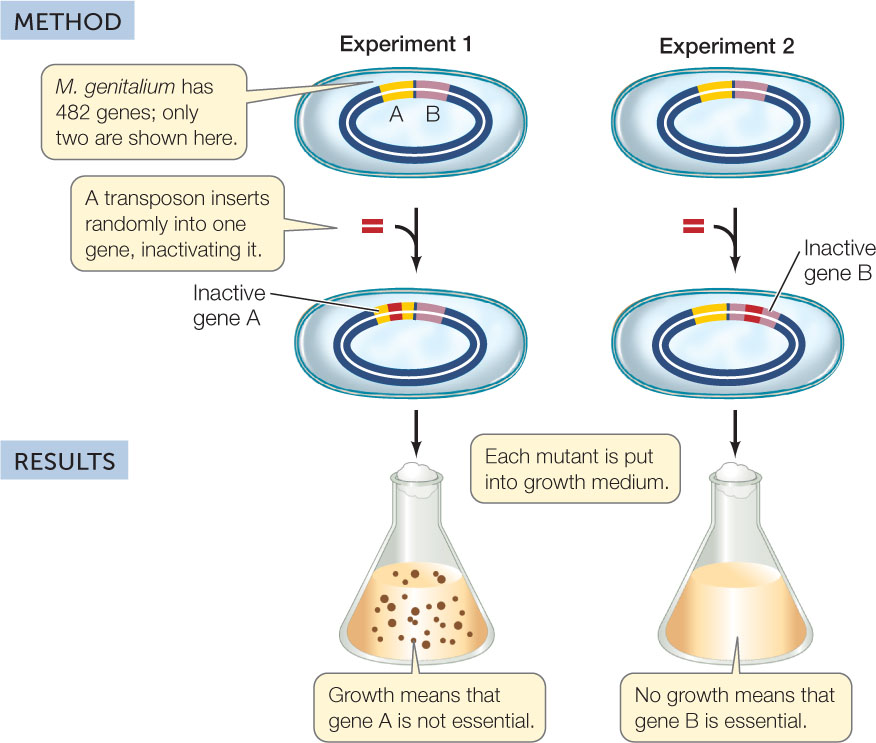
What about other genes? Researchers addressed this question using transposons as mutagens. When transposons in the bacterium were activated, they inserted themselves into genes at random, mutating and inactivating the genes (FIGURE 12.8). The mutated bacteria were tested for growth and survival, and DNA from interesting mutants was sequenced to find out which genes were mutated. The astonishing results of these studies suggested that only 382 of the 482 M. genitalium protein-coding genes were needed for survival in the laboratory!
Investigation
HYPOTHESIS
Only some of the genes in a bacterial genome are essential for cell survival.
CONCLUSION
If each gene is inactivated in turn, a “minimal essential genome” can be determined.
NALYZE THE DATA
The growth of M. genitalium strains with insertions in genes (intragenic regions) was compared with the growth of strains with insertions in noncoding (intergenic) regions of the genome:

- Explain these data in terms of genes essential for growth and survival. Are all of the genes in M. genitalium essential for growth? If not, how many are essential? Why did some of the insertions in intergenic regions prevent growth?
- If a transposon inserts into the following regions of genes, there might be no effect on phenotype. Explain why in each case:
- near the 3′ end of the coding region
- within a gene coding for rRNA
How does this affect your answer to the first question?
Go to LaunchPad for discussion and relevant links for all INVESTIGATION figures.
aC. Hutchison et al. 1999. Science 286: 2165–2169. J. I. Glass et al. 2006. Proceedings of the National Academy of Sciences USA 103: 425–430.
One application of the research might be to design organisms with specific uses. The next step toward that goal is to create an artificial genome and insert it into bacterial cells. As we described in the opening story of Chapter 4, this was recently accomplished, using a synthetic genome based on that of the bacterium Mycoplasma mycoides. This research has promise for making organisms with novel functions, such as the synthesis of plastics polymers or the ability to break down environmental pollutants.
CHECKpoint CONCEPT 12.2
- What are the characteristics of most prokaryotic genomes?
- Examine Table 12.1 and Figure 12.8. What gene functions would you predict are nonessential for M. genitalium as determined by transposon-mediated inactivation?
- You want to isolate a prokaryote that can live on discarded Styrofoam cups. Such an organism might live in a landfill where ground-up cups are discarded. How would you use metagenomics to identify such a bacterium?
- How would you show that the prokaryote’s ability to live on Styrofoam is essential, and that it cannot live in another environment?
243
The methods used to sequence and analyze prokaryotic genomes have also been applied to eukaryotic genomes, which we will examine next.