12.3 ISOLATION, IDENTIFICATION, AND SEQUENCING OF DNA FRAGMENTS
Watson and Crick’s discovery of the structure of DNA and knowledge of the mechanism of replication allowed biologists not only to understand some of life’s central processes, but also to create tools to study how life works. Biologists often need to isolate, identify, and determine the nucleotide sequence of particular DNA fragments. Such procedures can determine whether a genetic risk factor for diabetes has been inherited, whether blood at a crime scene matches that of a suspect, whether a variety of rice or wheat carries a genetic factor for insect resistance, and how closely two species of organisms are related. Many of the experimental procedures for the isolation, identification, and sequencing of DNA are based on knowledge of DNA structure and the physical properties of DNA. Others make use of the principles of DNA replication. In this section, we discuss how particular fragments of double-stranded DNA can be produced, how DNA fragments of different sizes can be physically separated, and how the nucleotide sequence of a piece of DNA can be determined.
12.3.1 The polymerase chain reaction selectively amplifies regions of DNA.
DNA that exists in the nuclei of your cells is present in just one copy per cell (in the case of the X or Y chromosome in males) or two copies per cell (in the case of the two X chromosomes in females and chromosomes other than the sex chromosomes). In the laboratory, it is very difficult to manipulate or visualize a sample containing just one or two copies of a DNA molecule. Instead, researchers typically work with many identical copies of the DNA molecule they are interested in. A common method for making copies of a piece of DNA is the polymerase chain reaction (PCR), which allows a targeted region of a DNA molecule to be replicated (or amplified) into as many copies as desired. PCR is both selective and highly sensitive, so it is used to amplify and detect small quantities of nucleic acids, such as HIV in blood-bank supplies, or to study DNA samples as minuscule as those left by a smoker’s lips on a cigarette butt dropped at the scene of a crime. The starting sample can be as small as a single molecule of DNA.
12-11
The principles of PCR are illustrated in Fig. 12.13. Because the PCR reaction is essentially a DNA synthesis reaction, it requires the same basic components used by the cell to replicate its DNA. In this case, the procedure takes place in a small plastic tube containing a solution that includes four essential components:
- Template DNA. At least one molecule of double-stranded DNA containing the region to be amplified serves as the template for amplification.
- DNA polymerase. The enzyme DNA polymerase is used to replicate the DNA.
- All four deoxynucleoside triphosphates. Deoxynucleoside triphosphates with the bases A, T, G, or C are needed as building blocks for the synthesis of new DNA strands.
- Two primers. Two short sequences of single-stranded DNA are required for the DNA polymerase to start synthesis. Enough primer is added so that the number of primer DNA molecules is much greater than the number of template DNA molecules.
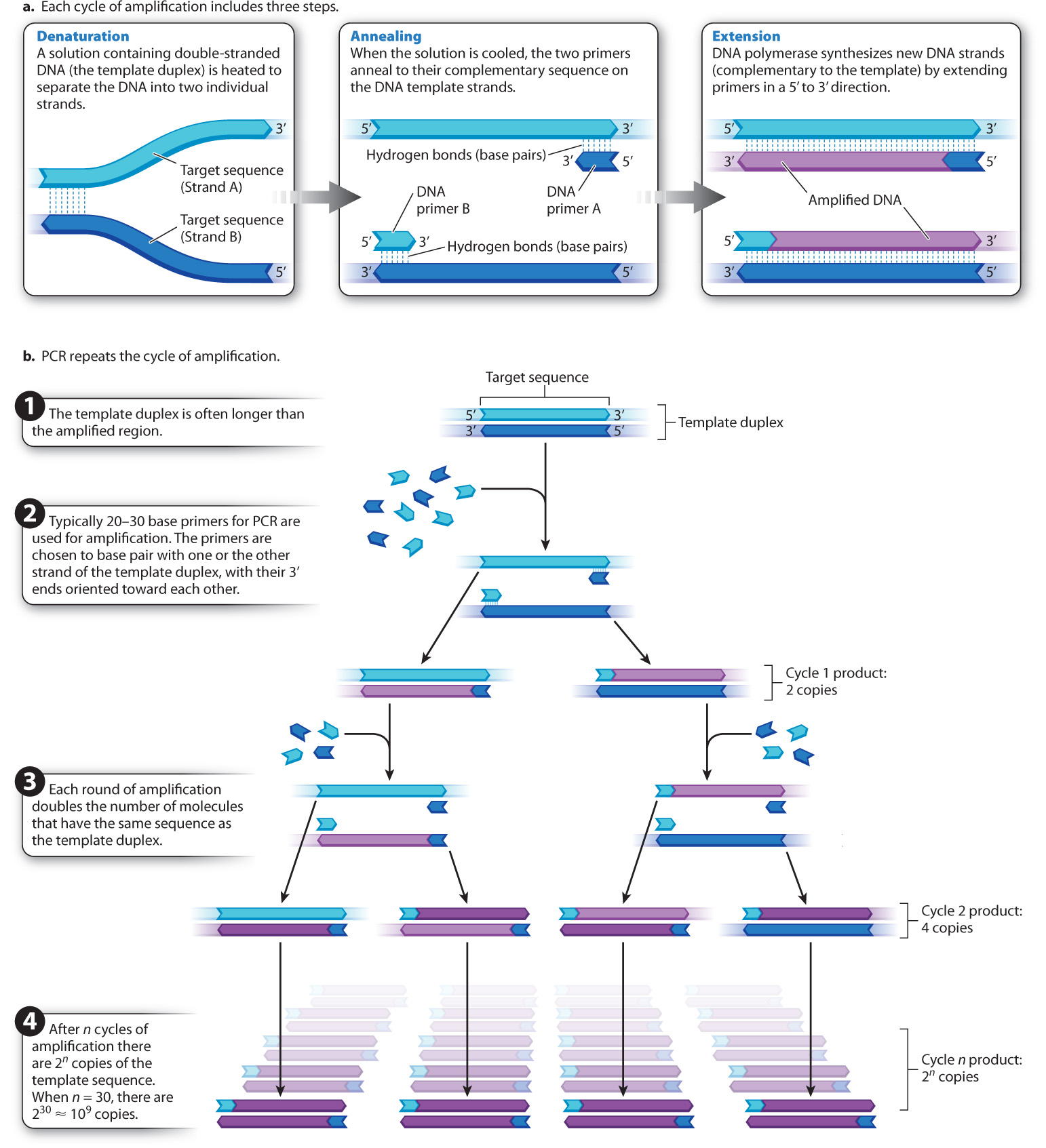
The primer sequences are oligonucleotides (oligos is the Greek word for “few”) produced by chemical synthesis and are typically 20–30 nucleotides long. Their base sequences are chosen to be complementary to the ends of the region of template DNA to be amplified. In other words, the primers flank the specific region of DNA to be amplified. The 3′ end of each primer must be oriented toward the region to be amplified, so that when DNA polymerase extends the primer, it creates a new DNA strand complementary to the targeted region. Because the 3′ ends of the primers both point toward the targeted region, one of the primers pairs with one of the template strands and the other pairs with the other template strand.
PCR creates new DNA fragments in a cycle of three steps, as shown in Fig. 12.13a. The first step, denaturation, involves heating the solution in the plastic tube to a temperature just short of boiling so that the individual DNA strands of the template duplex separate (or “denature”) as a result of the breaking of hydrogen bonds between the complementary bases. The second step, annealing, begins as the solution is cooled. Because of the great excess of primer molecules, the two primers bind (or “anneal”) to their complementary sequence on the DNA template (rather than two molecules of template coming back together). In the final step, extension, the solution is heated to the optimal temperature for DNA polymerase, and each primer is elongated (or “extended”) by means of the deoxynucleoside triphosphates.
After sufficient time to allow new DNA synthesis, the solution is heated again, and the cycle of denaturation, annealing, and extension is repeated over and over, as indicated in Fig. 12.13b, usually for 25–35 cycles. In each PCR cycle, the number of copies of the targeted fragment is doubled. The first round of PCR amplifies the targeted region into 2 copies, the next into 4, the next into 8, then 16, 32, 64, 128, 256, 512, 1024, and so forth. The doubling in the number of amplified fragments in each cycle justifies the term “chain reaction.”
Although PCR is elegant in its simplicity, the DNA polymerase enzymes from many species (including humans) irreversibly lose both structure and function at the high temperature required to separate the DNA strands. At each cycle, you would have to open the tube and add fresh DNA polymerase. This is possible, and in fact it was how PCR was done when the technique was first developed, but the procedure is time consuming and tedious. To solve this problem, we now use DNA polymerase from a bacterial species, Thermus aquaticus, that lives at the near–boiling point of water in natural hot springs such as those at Yellowstone National Park. This polymerase, called Taq polymerase, remains active at high temperatures. Once the reaction mixtures are set up, the entire procedure is carried out in a fully automated machine. The time of each cycle, temperatures, number of cycles, and other variables can all be programmed. The fact that DNA polymerase from a bacterium that lives in hot springs can be used to amplify DNA from any organism is further evidence for the conserved function and evolution early in the history of life of this enzyme.
12.3.2 Electrophoresis separates DNA fragments by size.
PCR amplification does not always work as theory says it should. Sometimes the primers are defective; sometimes they fail to anneal properly; sometimes they anneal to multiple sites and several different fragments are amplified. To determine whether or not PCR has yielded the expected product, a researcher must determine the size of the amplified DNA molecules. Usually, the researcher knows what the size of the correctly amplified fragment should be, making it possible to compare the expected size to the actual size.
12-12
12-13
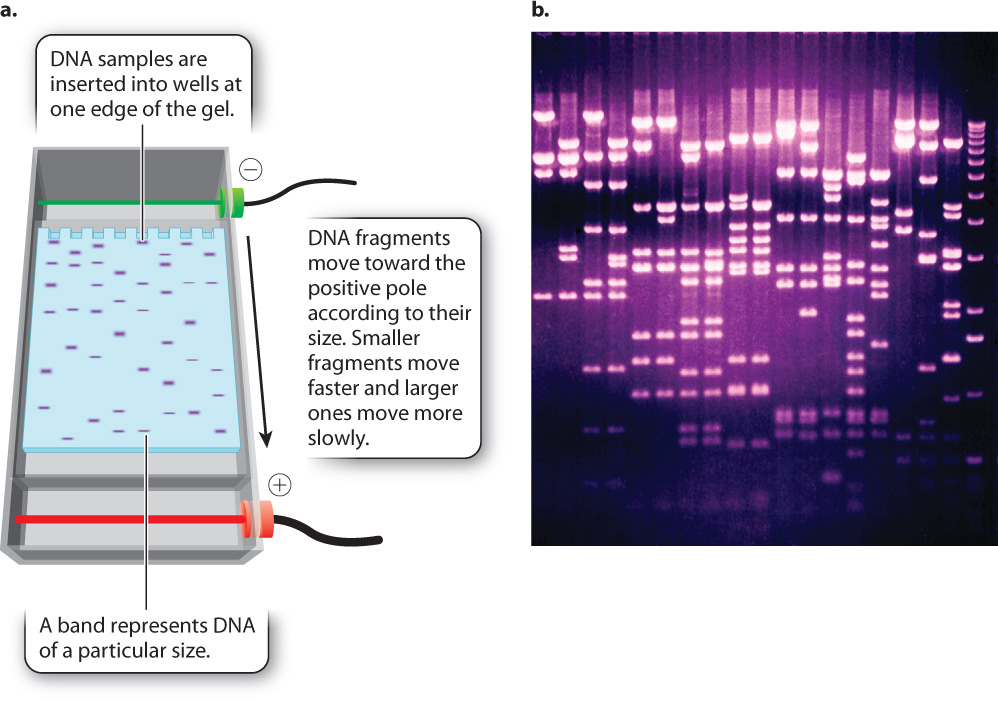
One way to determine the actual size of a DNA fragment is by gel electrophoresis (Fig. 12.14), a procedure in which DNA samples are inserted into slots or wells near the edge of a rectangular slab of porous material resembling solidified agar (the “gel”). The gel is then inserted into an apparatus and immersed in a solution that allows an electric current to be passed through it (Fig. 12.14a). Since fragments of double-stranded DNA are negatively charged because of the ionized phosphate groups along the backbone, the molecules move toward the positive pole of the electric field. The DNA molecules move according to their size. Short fragments pass through the pores of the gel more readily than large fragments, and so in a given interval of time short fragments move a greater distance in the gel than large fragments. The rate of migration is dependent only on size, not on sequence, and so all fragments of a given size move together at the same rate in a discrete band, which can be made visible by dyes that bind to DNA and fluoresce under ultraviolet light (Fig. 12.14b). A solution of DNA fragments of known sizes is usually placed in one of the wells, resulting in a series of bands, called a ladder, that can be used for size comparison.
Consider a PCR experiment with primers flanking a 250-base-pair (bp) length of DNA. PCR amplifies this region, theoretically creating many millions of copies of the 250-bp DNA fragment. To determine if the experiment worked as expected, a sample of the product is checked by gel electrophoresis. This sample is loaded into the well of the gel, a current is applied, and DNA fragments migrate to a position in the gel corresponding to their size, creating bands of DNA that are visualized with a dye. If a single band of 250 bp is seen on the gel, then the experiment worked as expected. Sometimes, however, the reaction might yield no bands, a single band of the incorrect size, or multiple bands. The researcher would then need to go back and investigate why the experiment did not work as expected.
Question Quick Check 2
KYsoo4WzWUKwnvhBgBX7E01sr8JXvuL21Jn2bBlG5Ft3QKI1VaTCia0amkmCTRDHhJJTHaGDEI27nO3YlHT+oxO/Egb3on8ZVzkIzm9LM2y65OHOspujUuOsWU88V8PpXpYacz9AesMKNyAsfSqvN6/xj2JqIco9s92/KwIfMUFr9swq9a1onQ6t/tv69Z/PBNmZZNtNKiRg8pFgJ1IMLHYiOG01i+t7Tlm+/R6JScZGF61N8YdE7e/APEF0pNAaXiTrWJU2P6FM0kkgYvpnm2bi88Y=Gel electrophoresis can separate DNA fragments produced by any means, not only PCR. Genomic DNA can be cut with certain enzymes and the resulting fragments separated by gel electrophoresis, as described in the next section. Similar procedures can also be used to separate protein molecules.
12.3.3 Restriction enzymes cleave DNA at particular short sequences.
In addition to amplifying segments of DNA, it is often useful to cut DNA at specific sites. Cutting DNA molecules allows pieces from the same or different organisms to be brought together in recombinant DNA technology, which is discussed in the next section. It also is a way to determine whether or not specific sequences are present in a segment of DNA, as techniques for cutting DNA depend on specific DNA sequences. Finally, cutting DNA allows whole genomes to be broken up into smaller pieces for further analysis, such as DNA sequencing.
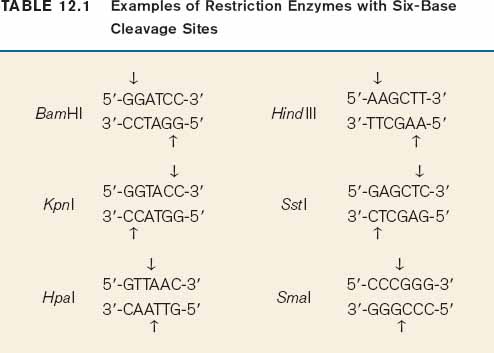
The method for cutting DNA makes use of a class of enzyme that recognizes specific, short nucleotide sequences in double-stranded DNA and cleaves the DNA at these sites. The enzymes are known as restriction enzymes, of which about 1000 different kinds have been isolated from bacteria and other microorganisms. The recognition sequences, called restriction sites, are typically four or six base pairs long, and most enzymes cleave double-stranded DNA at or near the restriction site. For example, the enzyme EcoRI has the following restriction site:
12-14
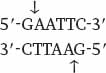
Wherever the enzyme finds this site in a DNA molecule, it cleaves each strand exactly at the position indicated by the vertical arrows. Note that the EcoRI restriction site is symmetrical: Reading from the 5′ end to the 3′ end, the sequence of the top strand is exactly the same as the sequence of the bottom strand. This kind of symmetry is called palindromic (that is, it reads the same in both directions) and is typical of restriction sites. Note also that the site of cleavage is not in the center of the recognition sequence. The cleaved double-stranded molecules therefore each terminate in a short single-stranded overhang. In this case the overhang is at the 5′ end, as shown below:
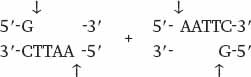
Table 12.1 shows more examples of restriction enzymes. Some cleave their restriction site to produce a 5′ overhang, others produce a 3′ overhang, and still others cleave exactly in the middle of their restriction site and leave blunt ends with no overhang. The standard symbols for restriction enzymes include both italic and roman letters. The italic letters stand for the species from which the enzyme is derived (E. coli in the case of EcoRI, Bacillus amyloliquefaciens in the case of BamHI), and the Roman letters designate the particular restriction enzyme isolated from that species.
When a particular restriction enzyme is used to break up a whole genome into smaller fragments, the specificity of restriction enzymes ensures that the DNA from each cell in an individual organism will yield the same set of fragments and that any particular DNA sequence will be present in a fragment of a particular size. If the genome is small enough that the number and sizes of the fragments are limited, the individual fragments can be visualized directly in a gel (Fig. 12.15). These fragments can then be extracted from the gel for further analysis or manipulation. For instance, the extracted fragments can be sequenced or ligated to other fragments.
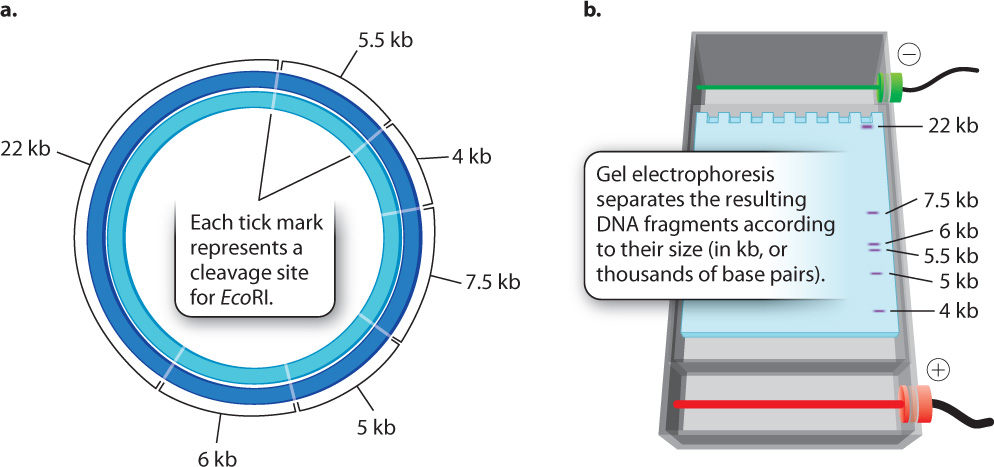
But most genomes are too large to give such a simple picture. For example, the human genome is cleaved into more than a million different fragments by EcoRI, and these fragments would appear as one big smear in a gel rather than as a series of discrete bands. In the next section, we discuss how individual bands containing a sequence of interest can be detected in such a gel.
12.3.4 DNA strands can be separated and brought back together again.
A researcher may wish to know whether a gene in one species is present in the DNA of a related species, but the nucleotide sequence of the gene is unknown. In such a case, techniques like PCR that require knowledge of the target DNA sequence cannot be used. Instead, the researcher can determine whether a DNA strand containing the gene in one species can base pair with a complementary sequence in a DNA strand from the other species. The base pairing of complementary single-stranded nucleic acids is known as renaturation or hybridization, and the process is the opposite of denaturation. In denaturation, the DNA strands in a duplex molecule are separated, and renaturation allows complementary strands to come together again. Denaturation and renaturation are also opposites in regard to the experimental conditions in which they occur. When a solution containing duplex DNA molecules is gradually heated to a sufficiently high temperature, the strands denature. When the solution is allowed to cool, the complementary strands renature.
12-15
Denatured DNA strands from one source can renature with DNA strands from a different source if their sequences are mostly complementary. Two very closely related sequences will have more perfectly matched bases, and thus more hydrogen bonds holding them together, than two sequences that are less closely related. The degree of base pairing between two sequences affects the temperature at which they renature—very closely related sequences renature at a higher temperature than less closely related sequences. Evolutionary biologists have used this principle to estimate the proportion of perfectly matched bases present in the DNA of different species, which is used as a measure of how closely species are related. In general, the more closely two species are related, the more similar their DNA sequences. Knowledge of species relatedness is important in many applications, including conservation, identifying endangered species, and tracing the evolutionary history of organisms.
Renaturation makes it possible to use a small DNA fragment as a probe. This fragment is usually attached to a light-emitting or radioactive chemical that serves as a label. The probe can be used to determine whether or not a sample of double-stranded DNA molecules contains sequences that are complementary to it. Any DNA fragment can be used as a probe, and a probe can be obtained in any number of ways, such as by chemical isolation of a DNA fragment, amplification by PCR, or nucleotide synthesis.
Let’s say you are interested in determining the number of copies of a particular DNA sequence or determining whether a given gene in human genomic DNA is intact. Recall that human and other genomic DNA cut with a restriction enzyme will yield a large number of DNA fragments that will look like a smear following gel electrophoresis. A labeled probe can be used to determine the size and number of a DNA sequence of interest in such a gel. The method is known as a Southern blot (Fig. 12.16) after its inventor, the British molecular biologist Edwin M. Southern.
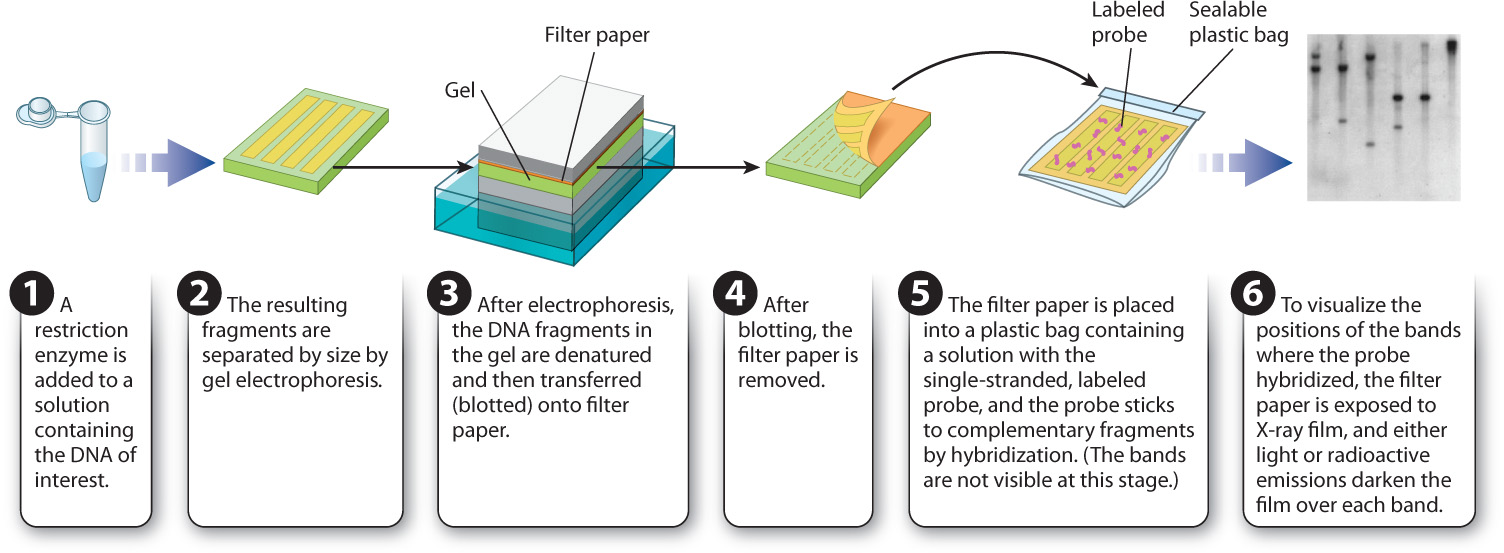
In a Southern blot, the DNA sample of interest is first digested with a restriction enzyme, and the resulting fragments are separated by size using gel electrophoresis. The DNA fragments within the gel are then denatured and transferred, or blotted, onto filter paper to which the DNA strands adhere. Each DNA fragment is transferred to the filter paper in the same position as it was in the gel. In the next step, the filter paper is covered with a solution containing the labeled probe DNA, and renaturation is allowed to proceed. The filter paper is then washed. Any probe DNA that has renatured with the strands stuck on the filter paper will remain in place, and any unbound probe DNA will be washed away. The final step is to overlay the filter paper with a sheet of X-ray film. Light emission or radioactivity from the labeled probe will cause the film to darken over the location of any fragment to which the probe binds.
The presence of a band or bands on the film therefore indicates the number and size of DNA fragments that are complementary to the probe. This information in turn tells you the sizes and the number of copies of a particular DNA sequence present in the starting sample.
12.3.5 DNA sequencing makes use of the principles of DNA replication.
The ability to determine the nucleotide sequence of DNA molecules has given a tremendous boost to progress in biological research. Techniques used to sequence DNA follow from our understanding of DNA replication. Consider a solution containing identical single-stranded template molecules of DNA, each in the process of directing the synthesis of a complementary daughter strand originating at a short primer sequence. The problem is to determine the nucleotide sequence of the template strand. A brilliant answer to this problem was developed by the English geneticist Frederick Sanger, an achievement rewarded with a share in the Nobel Prize in Chemistry in 1980. (It was his second Nobel; he had also been honored with the award in 1958 for his discovery of a method for determining the sequence of amino acids in a polypeptide chain.)
12-16
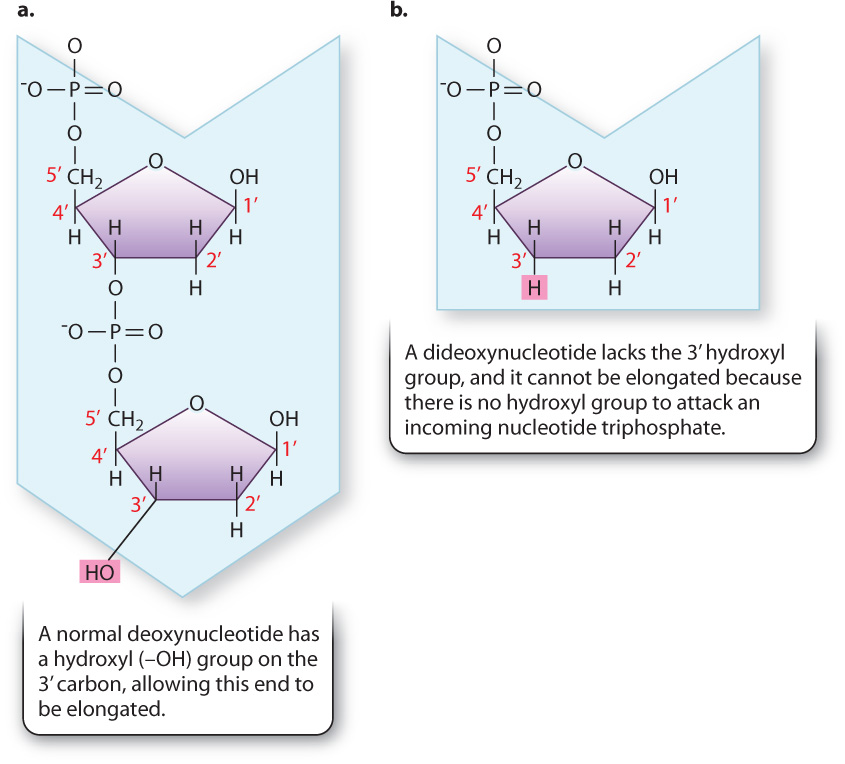
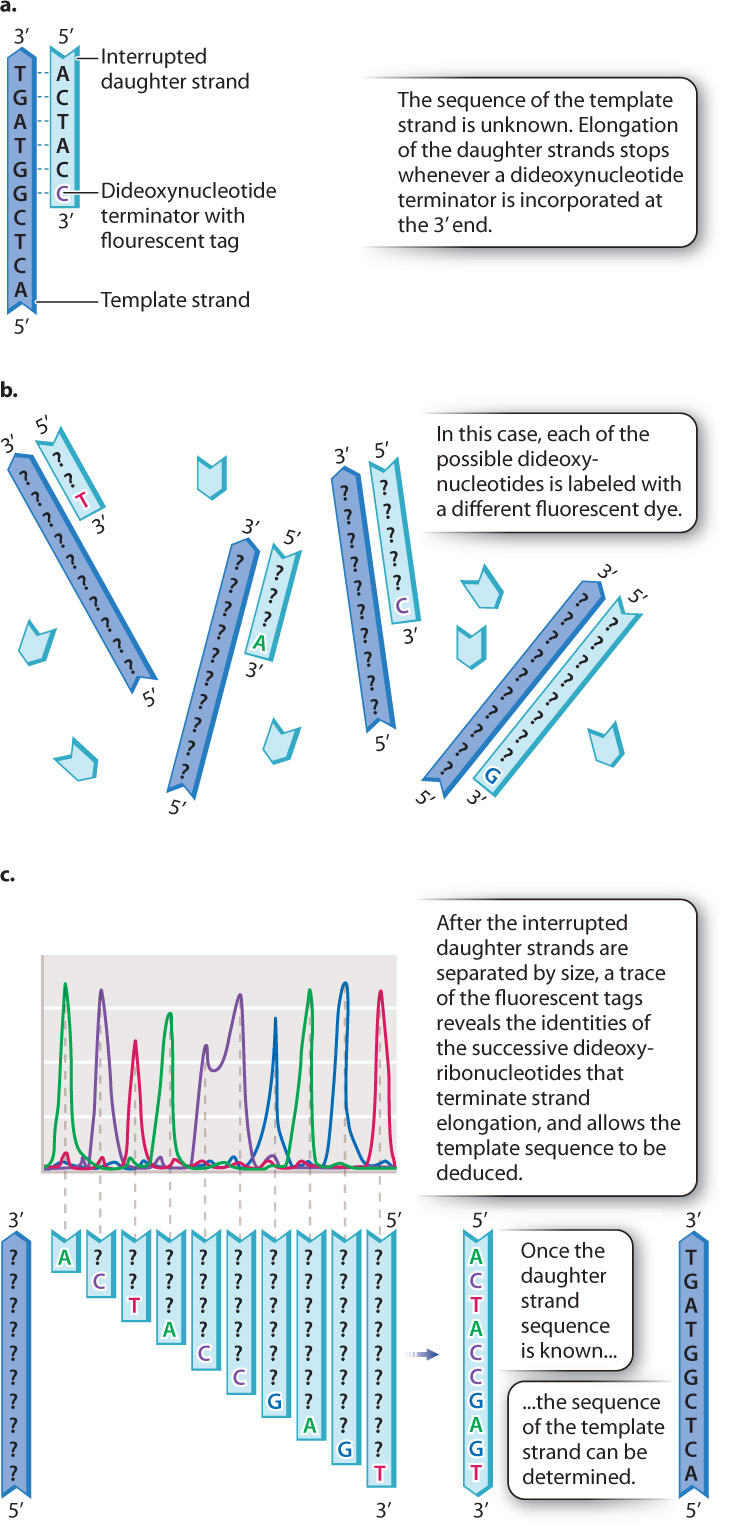
Recall that a free 3′ hydroxyl group is essential for each step in elongation because that is where the incoming nucleotide is attached (Fig. 12.17a). Making use of this fact, Sanger synthesized dideoxynucleotides, in which the 3′ hydroxyl group on the sugar ring is absent (Fig. 12.17b). Whenever a dideoxynucleotide is incorporated into a growing daughter strand, there is no hydroxyl group to attack the incoming nucleotide, and strand growth is stopped dead in its tracks (Fig. 12.18a). For this reason, a dideoxynucleotide is known as a chain terminator. By including a small amount of each of the chain terminators in a reaction tube along with all four normal nucleotides, a DNA primer, a DNA template, and DNA polymerase, Sanger was able to produce a series of interrupted daughter strands, each terminating at the site at which a dideoxynucleotide was incorporated.
Fig. 12.18b shows how the interrupted daughter strands help us to determine the DNA sequence by the procedure now called Sanger sequencing. In a tube containing dideoxy-A and all the other elements required for DNA replication, a strand of DNA is synthesized complementary to the template until, when it reaches a T in the template strand, it incorporates an A. Only a small fraction of the A nucleotides in the sequencing reaction are in the dideoxy form, so only a fraction of the daughter strands incorporate a dideoxy-A at that point, resulting in termination. The rest of the strands incorporate a normal deoxy-A and continue synthesis, although most of these will be stopped at some point farther along the line. Similarly, in a reaction containing dideoxy-C, DNA fragments will be produced whose sizes correspond to the positions of the Cs, and likewise for dideoxy-T and dideoxy-G.
Each of the four dideoxynucleotides is chemically labeled with a different fluorescent dye, as indicated by the different colors of A, C, T, and G in Fig. 12.18b, and so all four terminators can be present in a single reaction and still be distinguished. After DNA synthesis is complete, the daughter strands are separated by size using gel electrophoresis. The smallest daughter molecules migrate most quickly and therefore are the first to reach the bottom of the gel, followed by the others in order of increasing size. A fluorescence detector at the bottom of the gel “reads” the colors of the fragments as they exit the gel. What the scientist sees is a trace (or graph) of the fluorescence intensities, such as the one shown in Fig. 12.18c. The differently colored peaks, from left to right, represent the order of fluorescently tagged DNA fragments emerging from the gel. Thus, a trace showing peaks colored green-purple-red-green-purple-purple-blue-green-blue-red corresponds to a daughter strand having the sequence 5′-ACTACCGAGT-3′ (Fig. 12.18c). By use of the Sanger sequencing method, each sequencing reaction can determine the sequence of about 1000 nucleotides in the template DNA molecule.
Question Quick Check 3
zPf65m/d/5cyT+nJowI009SnWkhlm49g6xMzR86Xch5XX9AwzSyVTrZDpyYyFBYzgh6v09U9ZBQNiFfl1LDZ+S2lglbpYRk3vwd/tRJ9Gmt8jVCnukQfcLVEpX+S7Ea8rj4h4TcJg+2jVpnodfj5WWFjkFR1SzXOMFv2XaritSFNQNtt/aaAVu+5Z/PQbZIhLQEca/j1/P22RzWwmMHR+bCBJNSp+N5Xy8KY9m20c20=12.3.6 What new technologies will be required to sequence your personal genome?
Case 3 You, From A to T: Your Personal Genome
One of the high points of modern biology has been the determination of the complete nucleotide sequence of the DNA in a large number of species, including ours. The human genome and many others were sequenced by Sanger sequencing. This technique works well, but it takes time and is expensive for large genomes like the human genome. There have been many improvements in Sanger sequencing since it was first described, including the use of four-color fluorescent dyes to label the DNA fragments, capillary electrophoresis to separate the fragments, photocells to read the fluorescent signals automatically as the products run off the gel, and highly efficient enzymes. Together, these methods have increased the speed and decreased the cost of DNA sequencing considerably.
12-17
However, to be able to sequence everyone’s genome, including yours, we will require new technologies to bring down the cost further and to increase the speed of sequencing. Currently, there is a prize for anyone who can sequence a complete human genome for less than $1000. This $1000 genome will rely on next-generation sequencing technologies that get around some of the slower or more expensive steps of Sanger sequencing. For example, in a technique called pyrosequencing, the incorporation of a nucleotide into a growing DNA strand results in a pulse of light, allowing each base that is incorporated to be identified without having to separate DNA fragments on a gel. Another technique, called nanopore sequencing, passes individual DNA molecules through a pore in a membrane with a charge across it. As each nucleotide passes through the pore, it changes the charge across the membrane in a characteristic way, allowing identification.
Interest in large-scale DNA sequencing has stimulated the development of devices that increase scale and decrease cost. As in the development of computer hardware, emphasis is on making the sequencing devices smaller while increasing their capacity through automation. These miniaturized and automated approaches carry out what is often called massively parallel sequencing that can determine the sequence of hundreds of millions of base pairs in a few hours. These technologies will likely enable your own personal genome to be sequenced quickly and cheaply in the coming years.