13.2 GENOME ANNOTATION
A goal of biology is to identify all the component macromolecules in biological systems and to understand their individual functions and the ways in which they interact—their cellular organization, metabolism, growth, adaptation, reproduction, and other aspects of how life works. This research has practical applications: Increased understanding of the molecular and cellular basis of disease, for example, can lead to improved diagnosis and treatment.
The value of genome sequencing in identifying macromolecules is that the genome sequence contains, in coded form, the nucleotide sequence of all RNA molecules transcribed from the DNA as well as the amino acid sequence of all proteins. There is a catch, however. A genome sequence is merely an extremely long list of A’s, T’s, G’s, and C’s that represent the order in which nucleotides occur along the DNA in one strand of the double helix. (Because A in one strand is paired with T in the other and G is paired with C, knowing the sequence of one strand specifies the other.) The catch is that in multicellular organisms, not all of the DNA is transcribed into RNA, and not all of the RNA that is transcribed is translated into protein. Therefore, genome sequencing is just the first step in understanding the function of any particular DNA sequence. Following genome sequencing, the next step is to identify the locations and functions of the various types of sequence present in the genome.
13.2.1 Genome annotation identifies various types of sequence.
Genomes contain many different types of sequence, among them protein-coding genes. Protein-coding genes are themselves composed of different regions, including regulatory elements that specify when and where an RNA transcript will be produced, noncoding introns that are removed from the RNA transcript during RNA processing (Chapter 3), and protein-coding exons that contain the codons that specify the amino acid sequence of a polypeptide chain (Chapter 4). Genomes also contain coding sequences for RNAs that are not translated into protein (noncoding RNAs), such as ribosomal RNA, transfer RNA, and other types of small RNA molecule. Finally, much of the DNA in the genomes of multicellular organisms does not code for proteins or RNA at all, nor has it any other known function in the metabolism, physiology, development, or behavior of the organism.
Genome annotation is the process by which researchers identify the various types of sequence present in genomes. Genome annotation is essentially an exercise in adding commentary to a genome sequence that identifies which types of sequence are present and where they are located. It can be thought of as a form of pattern recognition, where the patterns are regularities in sequence that are characteristic of protein-coding genes or other types of sequence.
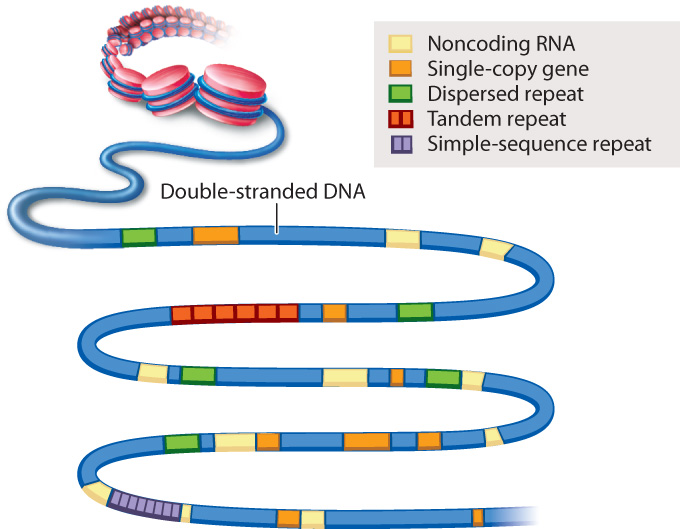
An example of genome annotation is shown in Fig. 13.3. Genes present in one copy per genome are indicated in orange. Most of these single-copy genes are protein-coding genes. The annotation of a single-copy gene typically specifies any nearby regulatory regions that control transcription, the intron–exon boundaries in the gene, and any known or predicted alternative forms in which the introns and exons are spliced (Chapter 3). Each single-copy gene is given a unique name and its protein product identified. Note in Fig. 13.3 that single-copy genes can differ in size from one gene to the next. The annotations in Fig. 13.3 also specify the locations of sequences that encode RNAs that are not translated into proteins, as well as various types of repeated sequence.
13-5
Small genomes such as that of HIV and other viruses can be annotated by hand, but for large genomes like the human genome, computers are essential. In the human genome, some protein-coding genes extend for more than a million nucleotides. A comparison helps convey the challenge of genome annotation. Roughly speaking, if the sequence of the approximately 3 billion nucleotides in a human egg or sperm was printed in normal-sized type, the length of the ribbon would stretch 4000 miles (6440 kilometers), about the distance from Fairbanks, Alaska, to Miami, Florida. By contrast, for the approximately 10,000 nucleotides in the HIV genome, the ribbon would extend a mere 70 feet (21 meters).
Genome annotation is an ongoing process because, as macromolecules and their functions and interactions become better understood, the annotations to the genome must be updated. A sequence that is annotated as nonfunctional today may be found to have a function tomorrow. For this reason, the annotation of certain genomes—including the human genome—will certainly continue to change.
13.2.2 Genome annotation includes searching for sequence motifs.
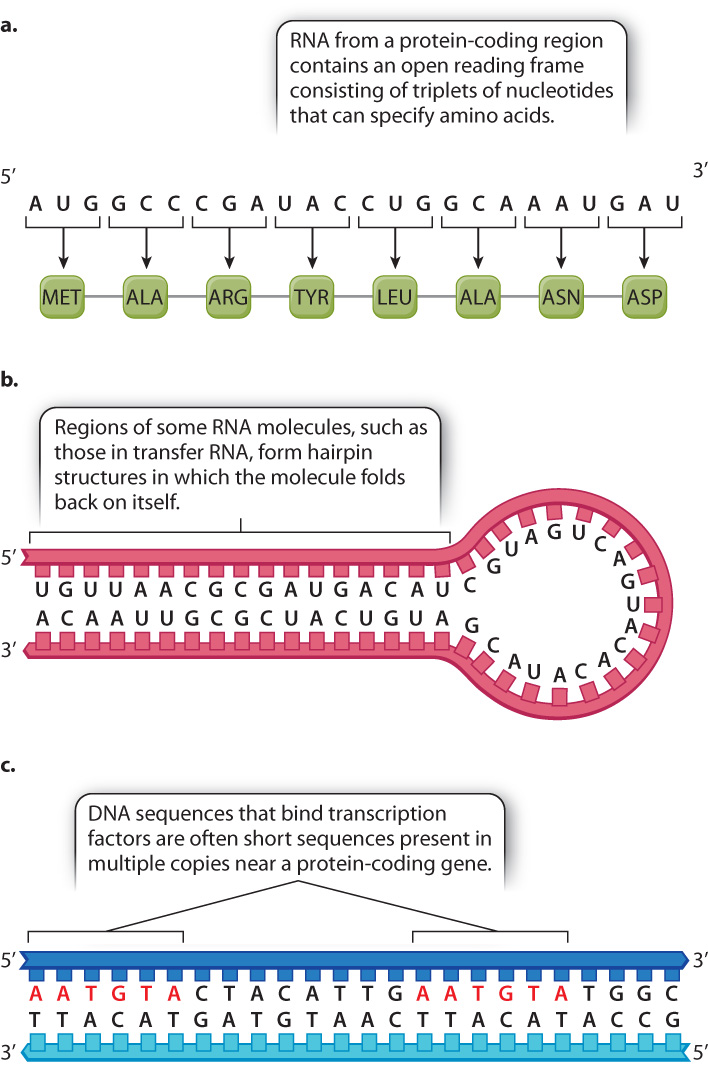
Because genome annotation is essentially pattern recognition, it begins with the identification of a sequence motif, a telltale sequence that indicates what type of sequence it is (Fig. 13.4). This sequence pattern can be found in the DNA itself, or in the RNA sequence inferred from the DNA sequence. Once identified, sequence motifs are typically confirmed by experimental methods.
An example of a sequence motif is an open reading frame (ORF) (Fig. 13.4a). The motif for an open reading frame is a long string of codons for amino acids with no stop codon. The presence of such a motif by itself is enough to annotate the DNA segment as potentially protein coding.
Fig. 13.4b shows another type of sequence motif, this one also present in a hypothetical RNA transcript inferred from the DNA sequence. The nucleotide sequence in one part of the RNA is complementary to that in another part, so the single-stranded molecule is able to fold back on itself and undergo base pairing to form a hairpin-shaped structure. Such hairpin structures are characteristic of certain types of RNA that function in gene regulation (Chapter 19). The DNA from which this RNA is transcribed would have complementary sequences on either end as well.
13-6
Some sequence motifs are detected directly in the double-stranded DNA. Fig. 13.4c shows two copies of a short sequence that are known binding sites for DNA-binding proteins called transcription factors (Chapter 3), whose binding to DNA initiates transcription. Transcription factor binding sites are often present in multiple copies and in either strand of the DNA. Sometimes they are located near the region of a gene where transcription is initiated because the transcription factor helps determine when the gene will be transcribed. However, they can also be located far upstream of the gene, downstream of the gene, or in introns, and so their identification is difficult.
13.2.3 Comparison of genomic DNA with messenger RNA reveals the intron–exon structure of genes.
In annotating an entire genome, researchers typically make use of information beyond the genome sequence itself. This information may include sequences of messenger RNA molecules that are isolated from various tissues or from various stages of development of the organism. Recall from Chapter 3 that messenger RNA (mRNA) molecules undergo processing and are therefore usually simpler than the DNA sequences from which they are transcribed—for example, introns are removed and exons are spliced together. The resulting mature mRNA therefore contains a long sequence of codons uninterrupted by a stop codon—in other words, an open reading frame. The open reading frame in an mRNA is the region that is actually translated into protein on the ribosome.
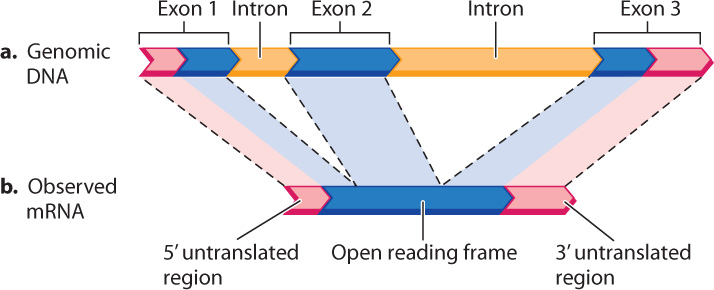
One aspect of genome annotation is the determination of which portions of the genome sequence correspond to sequences in mRNA transcripts. An example is shown in Fig. 13.5, which compares the DNA and mRNA for the β (beta) chain of hemoglobin, the oxygen-carrying protein in red blood cells. Note that the genomic DNA contains some sequences present in the mRNA, which correspond to exons, and some sequences that are not present in the mRNA, which correspond to introns. Comparison of mRNA with genomic DNA therefore reveals the intron–exon structure of protein-coding genes. In fact, introns were first discovered by comparing β-globin mRNA with genomic DNA.
13.2.4 An annotated genome summarizes knowledge, guides research, and reveals evolutionary relationships among organisms.
Genome annotation, which aims to identify all the functional and repeat sequences present in the genome, is an imperfect science. Even in a well-annotated genome, some protein-coding sequences or other important features may be overlooked, and occasionally the annotation of a sequence motif is incorrect.
Because researchers often have to rely on sequence motifs alone and not experimental data, their descriptions may be vague. For example, a common annotation in large genomes is “hypothetical protein.” In some cases, such as the genome of the malaria parasite, this type of annotation accounts for about 50% of the possible protein-coding genes. There is no hint of what a hypothetical protein may do or even whether it is actually produced, since it is determined solely by the presence of an ORF in the genomic sequence, and not by the presence of actual mRNA or protein. Other annotations might be “DNA-binding protein,” “possible hairpin RNA,” or “tyrosine kinase”—with no additional detail. In short, although some genome annotations summarize experimentally verified facts, many others are hypotheses and guides to future research.
Genome sequences contain information about ancestry and evolution, and so comparisons among genomes can reveal how different species are related. For example, the sequence of the human genome is significantly more similar to that of the chimpanzee than to that of the gorilla, indicating a more recent common ancestry of humans and chimpanzees (Chapter 1).
Analysis of the similarities and differences in protein-coding genes and other types of sequence in the genomes of different species is an area of study called comparative genomics. Such studies help us understand how genes and genomes evolve. They can also guide genome annotation because the sequences of important functional elements are often very similar among genomes of different organisms. Sequences that are similar in different organisms are said to be conserved. A sequence motif that is conserved is likely to be important even if its function is unknown, since it has changed very little over evolutionary time.
13.2.5 The HIV genome illustrates the utility of genome annotation and comparison.
HIV and related viruses provide an example of how genomes are annotated and how comparisons among genome sequences can reveal evolutionary relationships. A virus is a small infectious agent that contains a nucleic acid genome packaged inside a protein coat called a capsid. In some viruses, a lipid envelope surrounds the capsid. Viruses can bind to surface receptor molecules on cells of the host organism, enabling the viral genome to enter the cell. Infection of a host cell is essential to viral reproduction because viruses use cellular ATP and hijack cellular machinery to replicate, transcribe, and translate their genome in order to make more viruses.
13-7
Whereas the genome of all cells consists of double-stranded DNA, the genomes of viruses may be double-stranded DNA, single-stranded DNA, double-stranded RNA, or single-stranded RNA. The genome of HIV is single-stranded RNA. The sequence of the HIV genome identifies it as a retrovirus that replicates via a DNA intermediate that can be incorporated into the host genome. More narrowly, the sequence of the HIV genome groups it among the mammalian lentiviruses, so named because of the long lag between the initial time of infection and the appearance of symptoms (lenti- means “slow”).
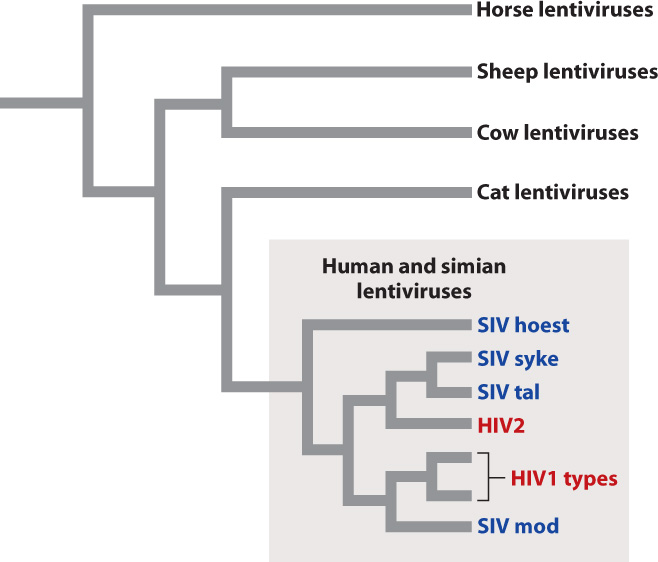
Fig. 13.6 shows the evolutionary relationships among a sample of lentiviruses, grouped according to the similarity of their genome sequences. The evolutionary tree shows that closely related viruses have closely related hosts. For example, cat lentiviruses are more closely related to one another than they are to cow lentiviruses. This observation implies that the genomes of the viruses evolve along with the genomes of their hosts. A second feature shown by the evolutionary tree is that human HIV originated from at least two separate simian viruses that switched hosts from simians (most likely chimpanzees) to humans.
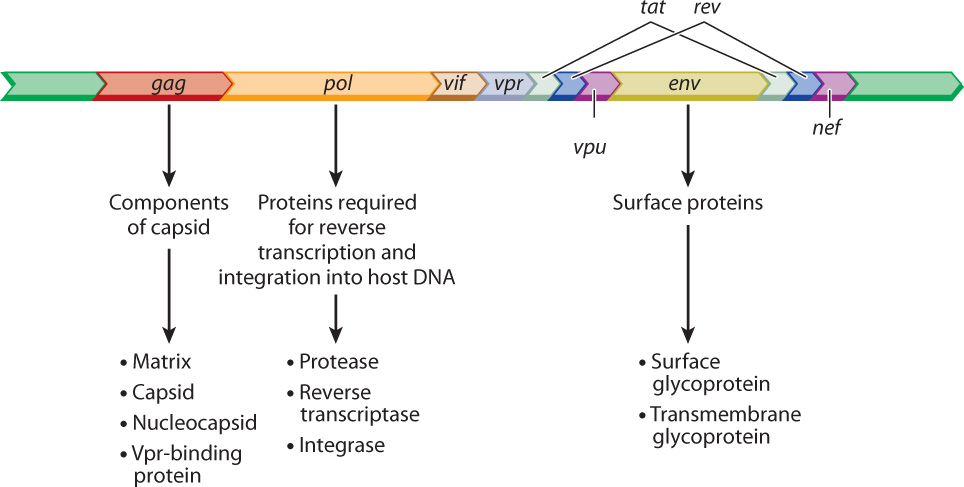
The annotated sequence of the HIV genome tells us a lot about the biology of this virus (Fig. 13.7). Many details are left out here, but the main point is to show the functional elements of HIV in the form of an annotated genome. The open reading frame denoted gag encodes protein components of the capsid, pol encodes proteins needed for reverse transcription of the viral RNA into DNA and incorporation into the host genome, and env encodes proteins that are embedded in the lipid envelope. The annotation in Fig. 13.7 also includes the genes tat and rev, encoding proteins essential for the HIV life cycle, as well as the genes vif, vpr, vpu, and nef, which encode accessory proteins that enhance virulence in organisms. Identification of the genes necessary to complete the HIV cycle is the first step to finding drugs that can interfere with the cycle and prevent infection.