19.1 CHROMATIN TO MESSENGER RNA IN EUKARYOTES
Gene regulation in multicellular eukaryotes leads to cell specialization: Different types of cell express different genes. The human body contains about 200 major cell types, and although for the most part they share the same genome, they look and function differently from one another because each type of cell expresses different sets of genes. For example, the insulin needed to regulate sugar levels in the blood is produced only by small patches of cells in the pancreas. Every cell in the body contains the genes that would lead to the production of insulin, but only in these patches of pancreatic cells are they expressed. In this section, we take a look at gene regulation as it occurs in eukaryotic cells, focusing on regulation at the level of DNA, chromatin, and mRNA.
19.1.1 Gene expression can be influenced by chemical modification of DNA or histones.
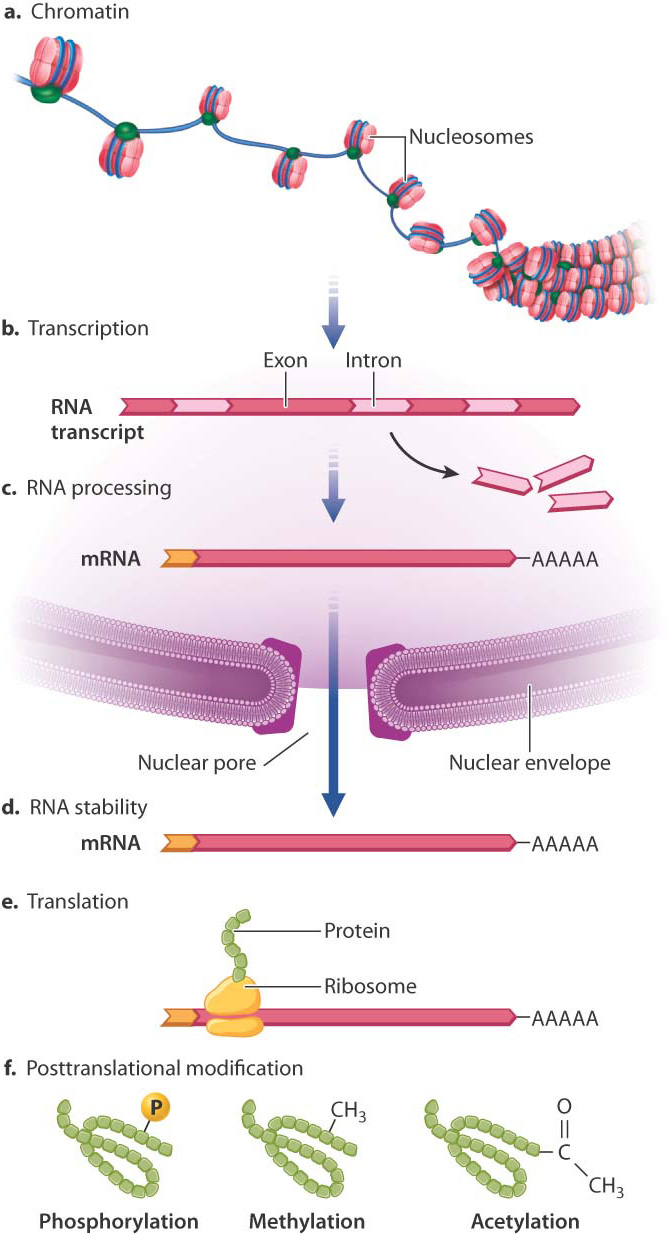
Fig. 19.1 shows the major places where gene regulation in eukaryotes can take place. The first level of control is at the chromosome, even before transcription takes place. The manner in which DNA is packaged in the nucleus in eukaryotes provides an important opportunity for regulating gene expression.
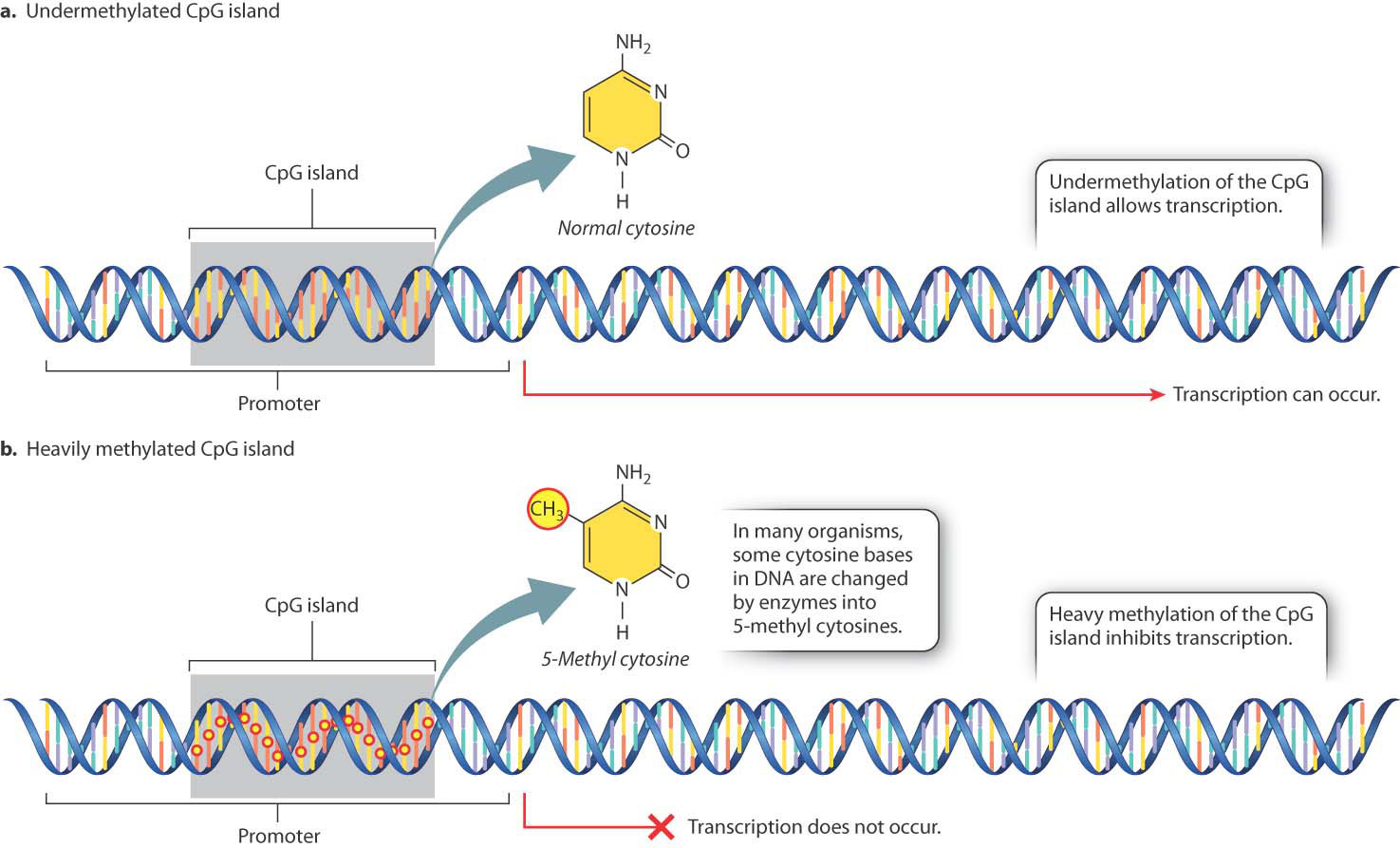
In many eukaryotic organisms, gene expression is affected by chemical modification of certain bases in the DNA (Fig. 19.2), the most common of which is the addition of a methyl group to the base cytosine. Methylation often occurs in cytosine bases that are adjacent to guanosine bases on a DNA strand. Such pairs of nucleotides are abbreviated “CpG” (the “p” represents the phosphate in the backbone of the DNA strand between the two nucleotides). In mammalian protein-coding genes, CpG sites are often clustered in small regions located in or near the promoter of the gene, the region where RNA polymerase and associated proteins bind to the DNA to initiate transcription. Such a cluster of CpG sites is known as a CpG island.
Some CpG sites in the genome are methylated and some are not. Methylated CpG sites are rarely seen in CpG islands near active genes (Fig. 19.2a), whereas transcriptional repression of a gene is often accompanied by heavy methylation of a nearby CpG island (Fig. 19.2b). The methylation state of a CpG island can change over time or in response to environmental cues, providing a way to turn genes on or off. Cells sometimes heavily methylate CpG islands of transposable elements or viral DNA sequences that are integrated into the genome, thus preventing the expression of genes in viruses and transposable elements (Chapter 14). In cancer cells, CpG island methylation often takes place, repressing genes that could restrict the cells’ growth.
DNA in eukaryotes is packaged as chromatin, a complex of DNA, RNA, and proteins that gives chromosomes their structure. Chromatin includes a thread of nucleosome particles in which about 150 base pairs of DNA are wrapped around each octamer of histone proteins. (Greater detail in the structure of chromatin and nucleosomes is depicted in Fig. 3.13.) When chromatin is in its coiled state, the DNA is not accessible to the proteins that carry out transcription. The chromatin must unravel to allow space for transcriptional enzymes and proteins to work. This is accomplished through chromatin remodeling, in which the nucleosomes are repositioned to expose different stretches of DNA to the nuclear environment. Chromatin remodeling is another level at which gene expression can be controlled.
19-3
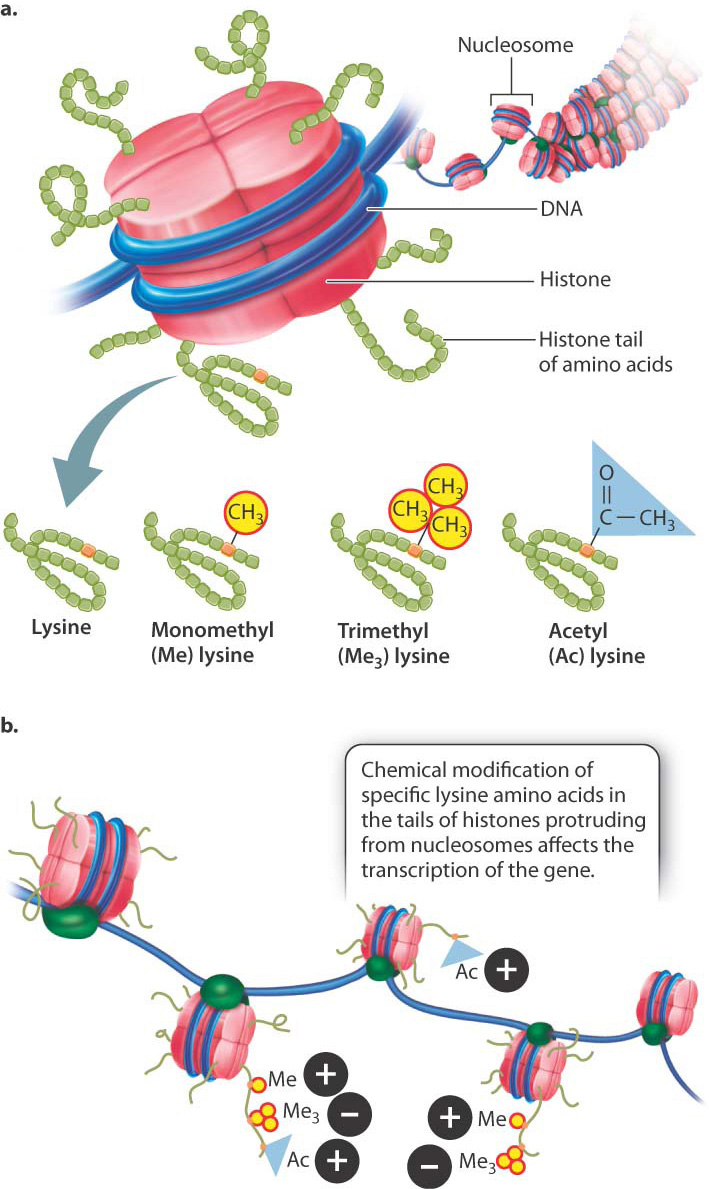
One way in which chromatin is remodeled is by chemical modification of the histones around which DNA is wound (Fig. 19.3). Modification usually occurs on histone tails, strings of amino acids that protrude from the histone proteins in the nucleosome (Fig. 19.3a). Individual amino acids in the tails can be modified by the addition (or later removal) of different chemical groups, including methyl groups (—CH3) and acetyl groups (—COCH3). Most often, methylation or acetylation occurs on the lysine residues of the histone tails.
The pattern of modifications of the histone tails is thought to constitute a histone code that affects chromatin structure and gene transcription (Fig. 19.3b). For example, methylation of lysine with a single methyl group and lysine acetylation are often associated with the activation of transcription (+ in Fig. 19.3b), whereas methylation of lysine with three methyl groups is often associated with repression of transcription (− in Fig. 19.3b). Modification of histones takes place at key times in development to ensure that the proper genes are turned on or off, as well as in response to environmental cues.
Together, these modifications of bases, changes to histones, and alterations in chromatin structure are often termed epigenetic, from the Greek epi- (“over and above,” “in addition to”) and genetic (“inherited”). That is, epigenetic mechanisms of gene regulation typically involve changes not to the DNA sequence itself but to the manner in which DNA is packaged. Epigenetic modifications can in some cases affect gene expression. They can be inherited through cell divisions, just as genes are, but are often reversible and responsive to changes in the environment.
19.1.2 Gene expression can be regulated at the level of an entire chromosome.
A striking example of an epigenetic form of gene regulation is the manner in which mammals equalize the expression of X-linked genes in XX females and XY males. For most genes, there is a direct relation between the number of copies of the gene (the gene dosage) and the level of expression of the gene. An increase in gene dosage increases the level of expression because each copy of the gene is regulated independently of other copies. For example, as we saw in Chapter 15, the presence of an extra copy of most human chromosomes results in spontaneous abortion because of the increase in the expression of the genes in that chromosome.
19-4
XX females and XY males have different numbers of X chromosomes. For genes contained in the X chromosome, the dosage of genes is twice as great in females as it is in males. However, the level of expression of X-linked genes is about the same in both sexes. These observations imply that the regulation of X-chromosomal genes is different in females and in males. The differential regulation is called dosage compensation.
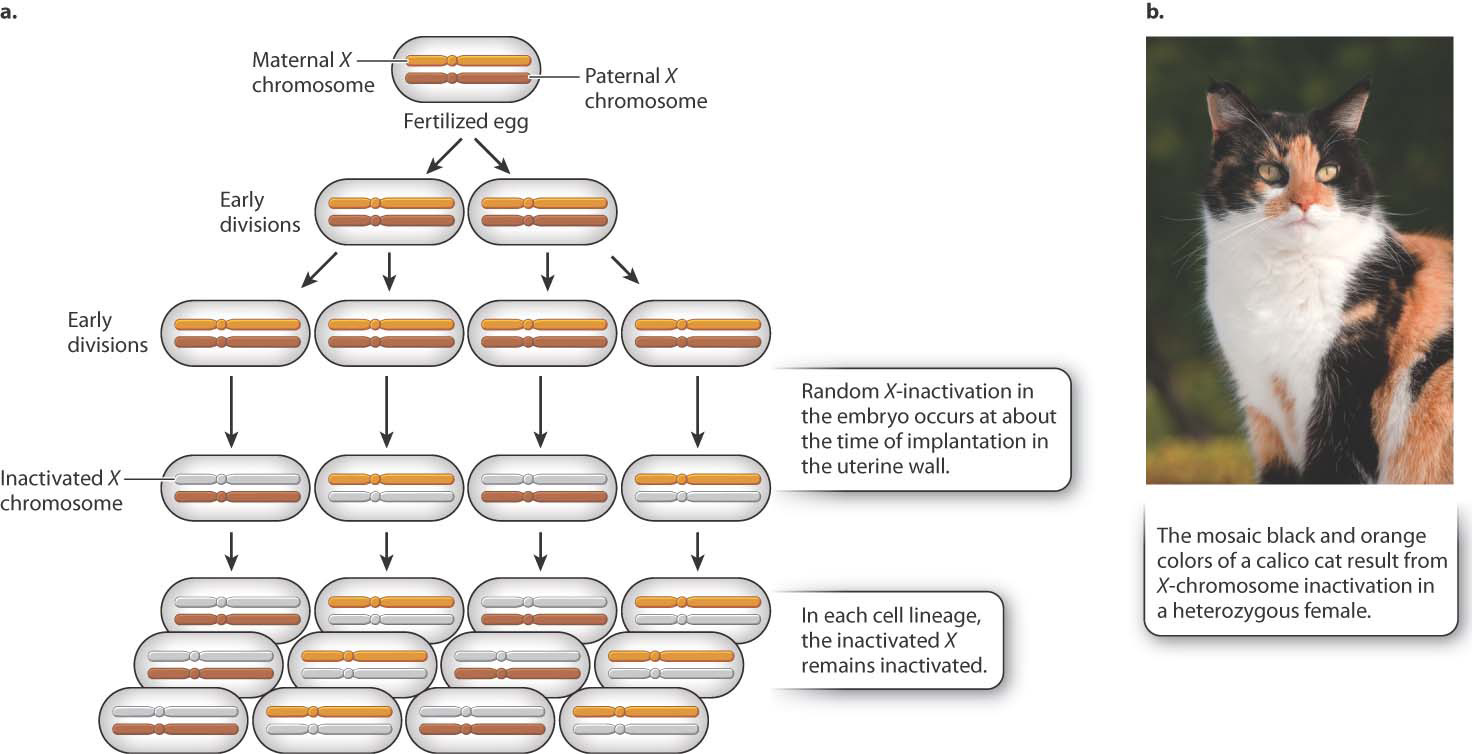
Different species have evolved different mechanisms of dosage compensation. In Drosophila fruit flies, males double the transcription of the single X chromosome to achieve equal expression compared to the two X chromosomes in females. In Caenorhabditis nematode worms, females decrease the transcription of both X chromosomes in females to one-half the level of the single X chromosome in males. In mammals, including humans, dosage compensation occurs through the inactivation of one X chromosome in each cell in females. This process, now known as X-inactivation, was first proposed by Mary F. Lyon in the early 1960s.
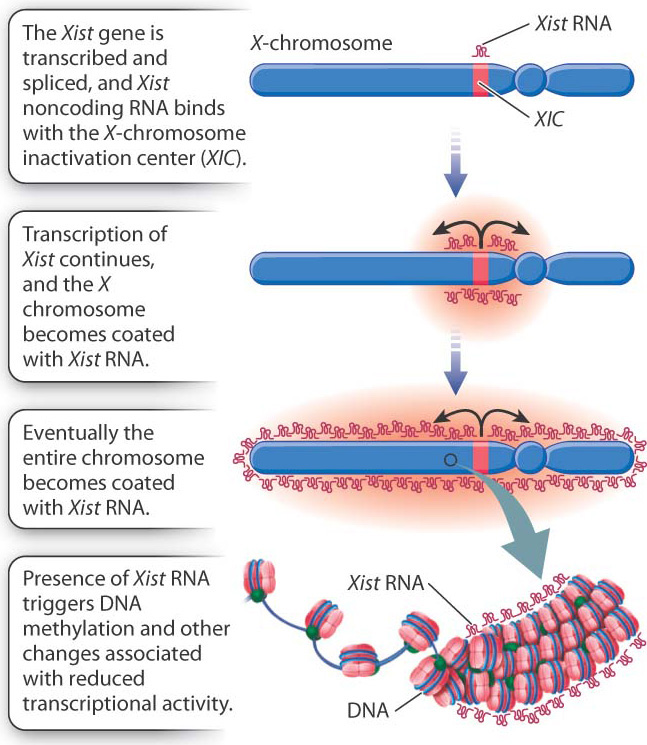
Soon after a fertilized egg with two X chromosomes implants in the mother’s uterine wall, one X chromosome is selected at random and inactivated (Fig. 19.4). In Fig. 19.4a, the inactive X chromosome is shown in gray. The inactive state persists through cell division, so in each cell lineage, the same X chromosome that was originally inactivated remains inactive. The result is that a normal female is a mosaic, or patchwork, of tissue. In some patches, the genes on the maternal X are expressed (and the paternal X is inactivated), whereas in other patches, the genes on the paternal X are expressed (and the maternal X is inactivated). The term “inactive X” is a slight exaggeration since a substantial number of genes are still transcribed, although usually at a low level.
As one argument for her X-inactivation hypothesis, Lyon called attention to calico cats, which are nearly always female (Fig. 19.4b). In calico cats, the orange or black fur colors are due to different alleles of a single gene in the X chromosome. In a heterozygous female, X-inactivation predicts discrete patches of orange and black, and this is exactly what is observed. (The white patches on a calico cat are due to an autosomal gene.)
How does X-inactivation work? Some of the details are still unknown, but the main features of the process are shown in Fig. 19.5. A key player is a small region in the X chromosome called the X-chromosome inactivation center (XIC), which contains a gene called Xist (X-inactivation specific transcript). The Xist gene is normally transcribed at a very low level, and the RNA is unstable, but in an X chromosome about to become inactive, Xist transcription markedly increases. The transcript undergoes RNA splicing, but it does not encode a protein. Xist RNA is therefore an example of a noncoding RNA, introduced in Chapter 3. Instead of being translated, the processed Xist RNA coats the XIC region, and as it accumulates, the coating spreads outward from the XIC until the entire chromosome is coated with Xist RNA. The presence of Xist RNA along the chromosome recruits factors that promote DNA methylation, histone modification, and other changes associated with transcriptional silencing.
19-5
19.1.3 Transcription is a key control point in gene expression.
While access to DNA and appropriate histone modifications are necessary for transcription, they are not sufficient. The molecular machinery that actually carries out transcription is also required once the template DNA is made accessible through chromatin remodeling and histone modification. The mechanisms that regulate whether or not transcription occurs are known collectively as transcriptional regulation (see Fig. 19.1b).
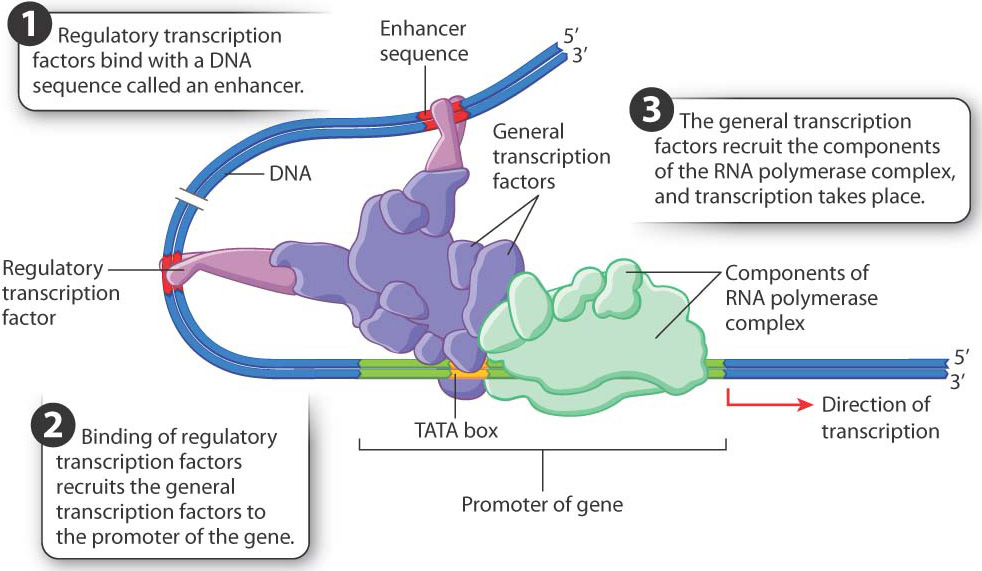
Transcriptional regulation in eukaryotic cells requires the coordinated action of many proteins that interact with one another and with DNA sequences near the gene. Let’s first review the basic process of transcription (Chapter 3). An important group of proteins are the general transcription factors. These proteins bind to the gene’s promoter, which is the region of a gene that recruits factors necessary to start transcription. The transcription factors are brought there by one of the proteins that binds to a short sequence called the TATA box, which is usually situated 25–30 nucleotides upstream of the nucleotide site where transcription begins. Once bound to the promoter, the transcription factors recruit the components of the RNA polymerase complex, which synthesizes the RNA transcript complementary to the template strand of DNA.
Where in the many steps of transcription initiation does regulation occur? The first place where transcription can be regulated is in the recruitment of the general transcription factors and components of the RNA polymerase complex (Fig. 19.6). Recruitment of these elements is controlled by proteins called regulatory transcription factors. Transcription does not occur if the regulatory transcription factors do not recruit the components of the transcription complex to the gene. Each regulatory transcription factor has two binding sites, one of which binds with a particular sequence in the DNA in or near a gene known as an enhancer (Fig. 19.6). A second binding site on the regulatory transcription factor recruits one or more general transcription factors to the promoter region. The general transcription factors then recruit the RNA polymerase complex, and transcription can begin (Chapter 3).
19-6
Hundreds of different regulatory transcription factors control the transcription of thousands of genes. A typical gene has several different types of enhancer sequence, each with its own regulatory transcription factor or set of transcription factors. Transcription takes place only when all the regulatory transcription factors are present and work together, as shown in Fig. 19.6. Transcription of a gene therefore depends on the presence of a particular combination of enhancers and their regulatory transcription factors, a type of regulation called combinatorial control.
Question Quick Check 1
HVmwR9sbcC67d7PQ6DGDHDSWo2/5DEH+7kiNHSxRTLfUMiknLLOL7z8V+suwDS0pxjaJL3O2GJUB806Hfe4fxEX84eAsgbXS6FMQBf9dCLGc87RIVwRa4pDByJj9JZbN6DYa0qEJEKUh+Vf/E8vx+mUCF6W4mrWtzQtP9Q98rAc1aHNvNjt0oTC1SaoCZfkcfKkTxJY0fwaXiU9dgPq+8hlHRlIX5tbD7TRhgVWgdudfUV3XHrAo+EAlqrhBNRusQ1I/WSNf+1l+qj1Kl5NwkDd9ZncywBa+Gzq5+TIbnXtSdxIRXFFNS8pS7KPDBwCxmqhQu+aAC/G47zXKZYcVozp50J3bmapgVVqBPpIrFDGCPuNMId4g/0yqZ80lSoTNSvWJczANNPC2cdEfnDzsGBFkqESOLLmG7LW7G0H7dWqDwYFTYAherNxVO857UBK4tXppYNTDa1OC2wxWqbUG8ZqUYFxr2ao/soQi1J71ZlRBaP0LQOkwT+4OF43o9NnBxnrbCalWH8cobFOGTH9Hyg==19.1.4 RNA processing is also important in gene regulation.
A great deal happens in the nucleus after transcription takes place. The initial transcript, called the primary transcript, undergoes several types of modification, collectively called RNA processing (Chapter 3), that includes the addition of a nucleotide cap to the 5′ end and a string of tens to hundreds of adenosine nucleotides to the 3′ end to form the poly(A) tail. These modifications are necessary for the RNA molecule to be transported to the cytoplasm and recognized by the translational machinery, and they also help to determine how long the RNA will persist before being degraded. RNA processing is therefore an important point where gene regulation can occur (see Fig. 19.1c).
In eukaryotes, the primary transcript of many protein-coding genes is far longer than the messenger RNA ultimately used in protein synthesis. The long primary transcript consists of regions that are retained in the messenger RNA (the exons) interspersed with regions that are excised and degraded (the introns). The introns are excised during RNA splicing (Chapter 3). The exons are joined together in their original linear order to form the processed messenger RNA.
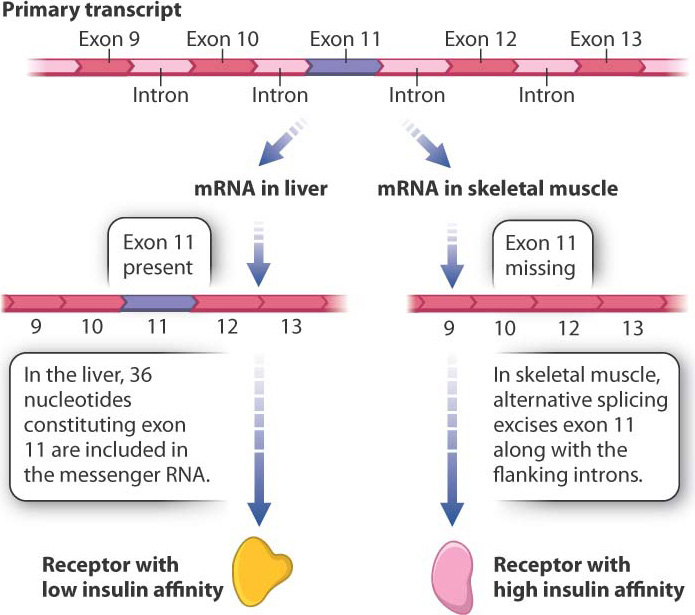
RNA splicing provides an opportunity for regulating gene expression because the same primary transcript can be spliced in different ways to yield different proteins in a process called alternative splicing. This process takes place because what the spliceosome—the splicing machinery—recognizes as an exon in some primary transcripts it recognizes as part of an intron in other primary transcripts. The alternative-splice forms may be produced in the same cells or in different types of cell. Alternative splicing accounts in part for the observation that we produce many more proteins than our total number of genes. By some estimates, over 90% of human genes undergo alternative splicing.
Fig. 19.7 shows the primary transcript of a gene encoding an insulin receptor found in humans and other mammals. During RNA splicing in liver cells, exon 11 is included in the messenger RNA, and the insulin receptor produced from this messenger RNA has low affinity for insulin. In contrast, in cells of skeletal muscle, the 36 nucleotides of exon 11 are spliced out of the primary transcript along with the flanking introns. The resulting protein is 12 amino acids shorter, and this form of the insulin receptor has high affinity for insulin. The different forms of the protein are important: The higher sensitivity of muscle cells to insulin enables them to absorb enough glucose to fulfill their energy needs.
19-7
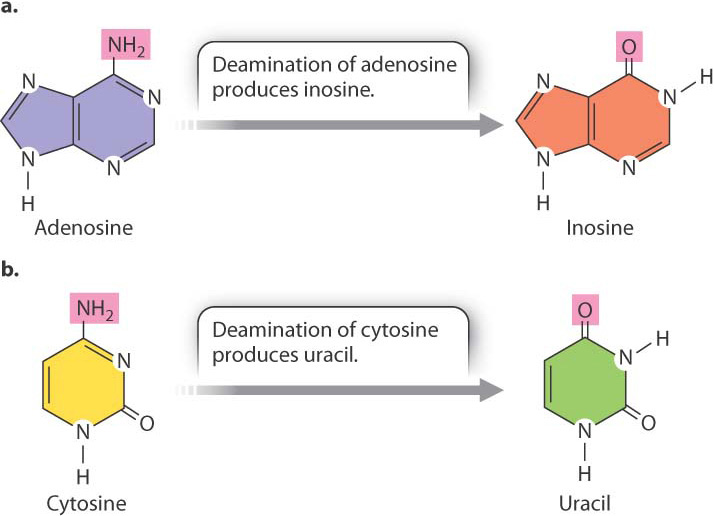
Some RNA molecules can become a substrate for enzymes that modify particular bases in the RNA, thereby changing its sequence and what it codes for. This process is known as RNA editing (Fig. 19.8). One type of editing enzyme (Fig. 19.8a) removes the amino group (—NH2) from adenosine and converts it to inosine, a base that in translation functions like guanosine. Another enzyme (Fig. 19.8b) removes the amino group from cytosine and converts it to uracil. In the human genome, hundreds if not thousands of transcripts undergo RNA editing. In many cases, not all copies of the transcript are edited, and some copies may be edited more extensively than others. The result is that transcripts from the same gene can produce multiple types of proteins even in a single cell.
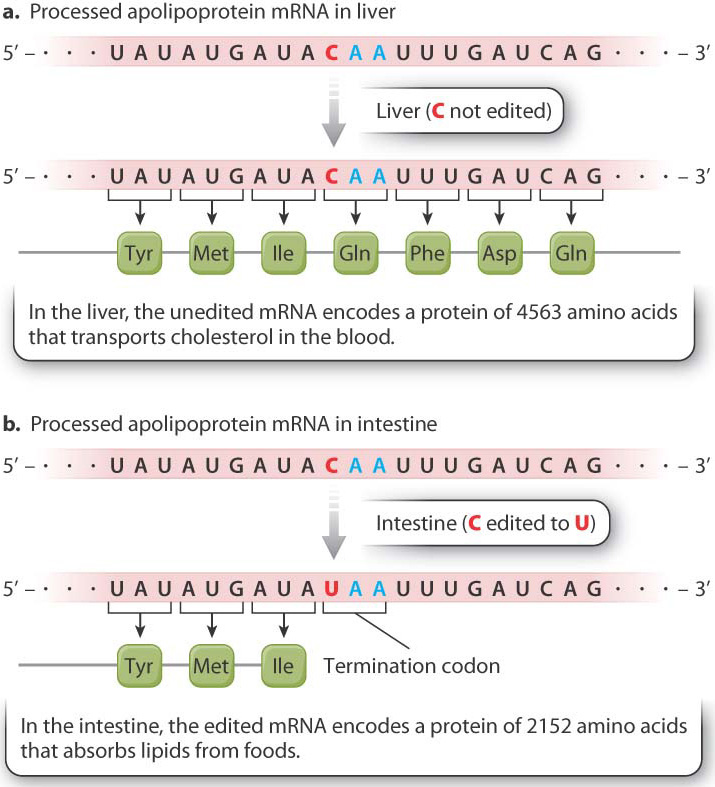
Transcripts from the same gene may undergo different editing in different cell types. An example of tissue-specific RNA editing is shown in Fig. 19.9. The mRNA fragments show part of the coding sequence for apolipoprotein B. The unedited mRNA in the liver (Fig. 19.9a) is translated into a protein that transports cholesterol in the blood. In contrast, RNA editing of the message occurs in the intestine (Fig. 19.9b). The cytosine nucleotide in codon 2153 is edited to uracil. The edited codon is UAA, which is a stop codon. Translation therefore terminates at this point, releasing a protein only about half as long as the liver form. This shorter form of the protein helps the cells of the intestine absorb lipids from the foods we eat.
19-8