26.4 THE DIVERSITY OF BACTERIA
Traditionally, bacterial taxonomy was a laborious process. Microbiologists grew bacteria in beakers or on petri dishes, carefully separating and enriching individual populations until only one type of bacterium—that is, a pure culture—was present. Species were identified on the basis of details of cell structure and division observed under the microscope, metabolic capabilities established through experiments and reactions to chemical stains or antibiotics. By the 1980s, a few thousand species had been described in this way.
With molecular sequencing technologies, however, bacterial species are now characterized by DNA sequences, especially the genes for the small subunit of ribosomal RNA (rRNA). A novel application of sequencing technology paved the way for modern studies of bacterial diversity and ecology. Microbiologists reasoned that the same procedures used to extract, amplify, and sequence genes from pure cultures in the lab could be used to study bacteria in nature. That is, extracting and reading gene sequences directly from soil or seawater could reveal the diversity of the microbial communities in these environments.
The results were stunning. Most bacteria in nature—perhaps 99% or more—are species that have not been cultured in the lab. Indeed, species well characterized by studies of pure cultures commonly represent only minor components of natural communities. To give just one illustration, a bacterium called SAR11 was originally identified on the basis of gene sequences amplified from seawater. SAR11 caught the attention of microbiologists because it makes up about a third of all sequences identified in the samples from the surface ocean. How could we be ignorant of a bacterium that must be among the most abundant organisms on Earth? The answer is simple: No one had cultured it. Recently, SAR11 has been cultured. Formally described as Pelagibacter ubique (ubique is Latin for “everywhere”), this bacterium is now known to be a heterotroph that plays an important role in recycling organic molecules dissolved in the sea.
More recently, the application of shotgun sequencing techniques (Chapter 13) to the environmental sampling of genomes has allowed microbiologists to begin matching uncultured populations with specific metabolisms. That is, we need no longer be content to know from molecular sequences what is living in a given environment. Now we can begin to understand what the different microorganisms are doing there. For this reason, another revolution in our understanding of the microbial world lies just around the corner (Fig. 26.13).
FIG. 26.13: How many kinds of bacterium live in the oceans?
BACKGROUND The oceans are full of bacteria—by some estimates, the sea harbors more than 1029 bacterial cells. How diverse are these bacterial communities?
METHOD 1 Different types of bacterium can be recognized by the unique nucleotide sequences of their genes. In fact, individual types of bacterium can be identified on the basis of nucleotide sequence in a relatively small region of the gene for the small subunit of ribosomal RNA (rRNA). This molecular “barcode” can be sequenced quickly and accurately for samples of DNA drawn from the environment.
Using this strategy, scientists associated with the International Census of Marine Life obtained samples of seawater and seafloor sediment from deep-ocean environments. In the laboratory, the rRNA sequences were amplified by PCR, separated by gel electrophoresis, and sequenced, thus providing a library of tags that document bacterial diversity.
METHOD 2 In another set of experiments, scientists collected samples of seawater from the Sargasso Sea, and from these they amplified whole genomes—more than a billion nucleotides’ worth. They used small-subunit rRNA and other genes to characterize species richness, and also characterized a large assortment of additional sequence data to understand genetic and physiological diversity.
RESULTS Both surveys found that the bacterial diversity of marine environments is much higher than had been thought. The barcode survey (method 1) found as many as 20,000 distinct types of bacteria in a single liter of seawater and estimated that as many as 5–10 million different microbes may live in the world’s oceans. Some of these bacteria are abundant and widespread, but most are rare. The genomic survey (method 2) of the Sargasso Sea found more than 1800 distinct bacteria in sea-surface samples and, remarkably, identified more than 1.2 million previously unknown genes within these genomes.
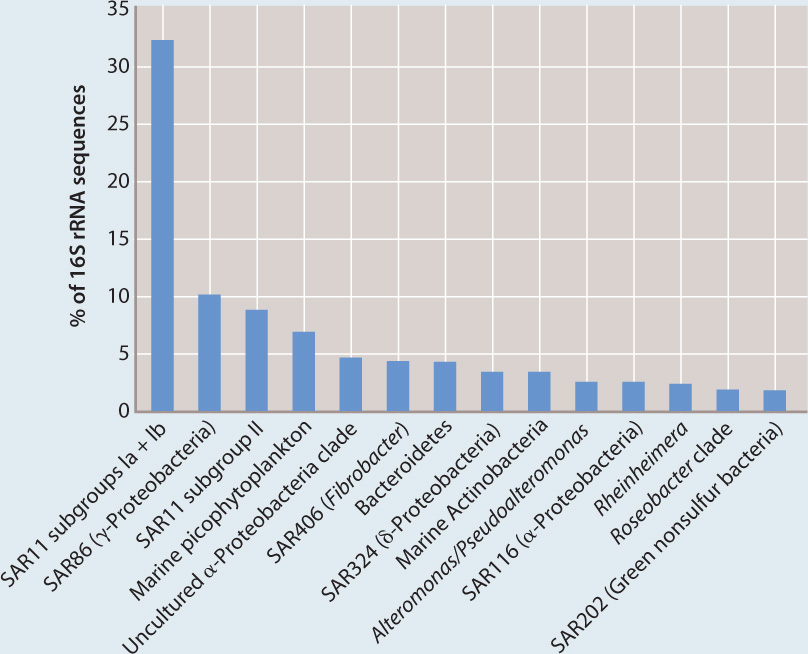
Relative abundance of different bacterial groups in samples from the Sargasso Sea, based on percent representation of small subunit rRNA sequences. Note the abundance of SAR11, only recently characterized.
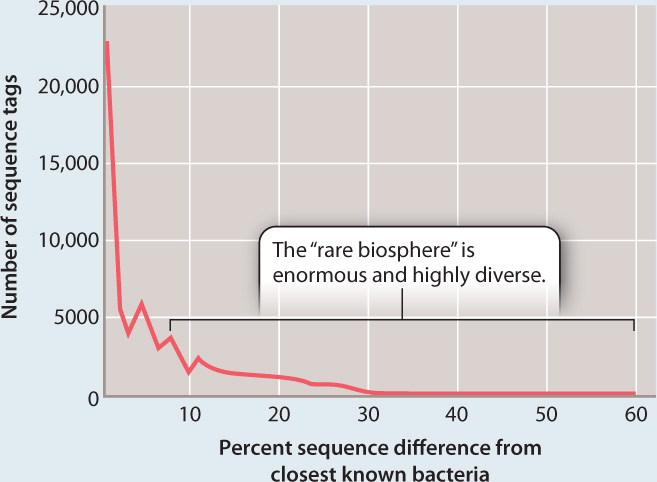
Computer-aided comparison of sequences from the world’s oceans shows that about 75% of all sequences collected are similar to other sequences in the library of sequence tags. However, 25% are distinctive, forming a rare but enormously diverse pool of bacterial types.
CONCLUSION The bacterial diversity of the oceans is huge and still largely unexplored. Both barcode and genomic surveys are continuing, and promise a more nearly complete accounting of marine diversity. Together, these surveys will help us understand how marine bacteria function and how they sort into communities.
SOURCES Venter, J. C., et al. 2004. “Environmental Genome Shotgun Sequencing of the Sargasso Sea.” Science 304:66–74; Sogin, M. L., et al. 2006. “Microbial Diversity in the Deep Sea and the Underexplored ‘Rare Biosphere’” Proceedings of the National Academy of Sciences USA 103:12115–12120.
26.4.1 Bacterial phylogeny is a work in progress.
In Chapter 23, we introduced the concept of molecular phylogeny, in which gene sequences are compared to draw conclusions about evolutionary relatedness among populations in nature. Gene sequencing made it possible to test traditional bacterial groupings based on form and physiology. Some traditionally defined groups—for example, the cyanobacteria— passed the test and stand today as a well-defined bacterial group. Many, however, did not.
Molecular sequence comparisons bring enormous new possibilities to the study of prokaryotic diversity and evolution, but they introduce new problems as well. One set of problems arises because major groups of Bacteria and Archaea separated from one another billions of years ago. Inherited sequence similarities can be masked by continuing molecular evolution within groups. That is, specific nucleotides in gene sequences may change not once but multiple times through a long evolutionary history, erasing molecular features that were once the same because of inheritance from a common ancestor. Also, in some groups rates of sequence evolution appear to be higher than in others. As a result, these groups have especially divergent sequences and so may misleadingly fall toward the bottoms of phylogenetic trees.
Another problem arises because of horizontal gene transfer. Phylogenies may falsely group distantly related bacteria because they contain genes passed on by conjugation, transformation, or transduction. Statistical analyses of complete bacterial genomes suggest that at least 85% of all genes in bacterial genomes have been transferred horizontally at least once. Such apparently rampant gene exchange might well doom attempts to build a bacterial tree of life, and some biologists argue that bacterial evolution is more properly depicted as a complex bush with many connecting branches (Fig. 26.14).
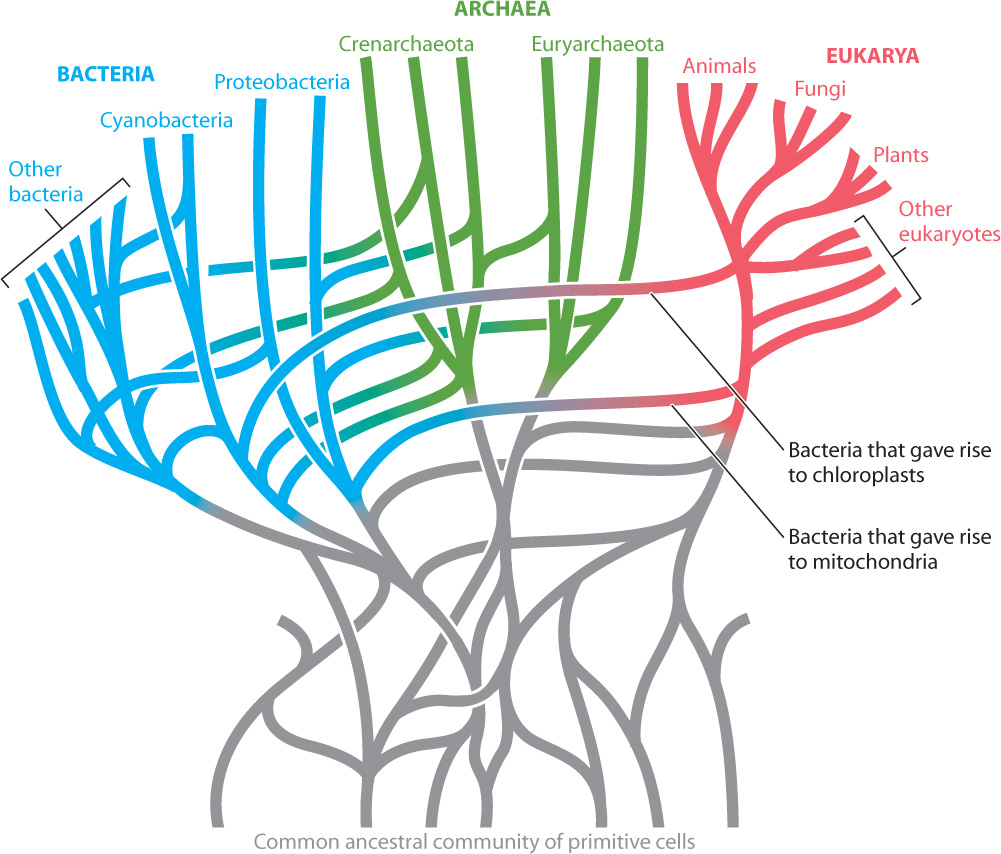
26-13
26-14
Other microbiologists are more optimistic. Small-subunit rRNA genes, for example, show little evidence of horizontal transfer and so may faithfully record the evolutionary history of the organisms in which they occur. Nonetheless, we need to be cautious when using bacterial trees to study the evolution of specific metabolic or physiological characteristics. Nitrogen fixation, photosystems, drug tolerance—such features can and do depend on genes known to jump from branch to branch on the tree.
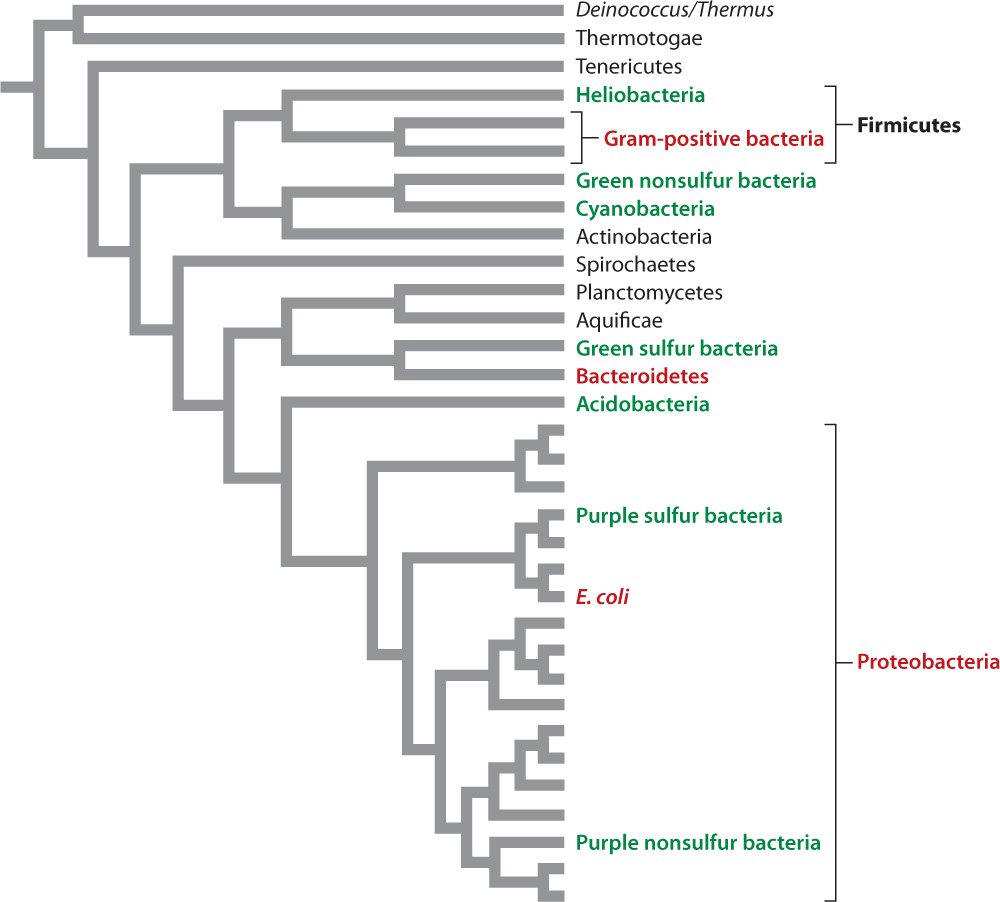
At present, molecular signatures have been extremely useful in identifying major groups of bacteria, but less clear in establishing branching order among these groups. About 50 major groups of bacteria (each one very roughly equivalent to a eukaryotic phylum) have been recognized. Nearly half of these groups are not represented in pure cultures. Rather, they have been identified only by the cloning of genes from environmental samples. To support our discussion of bacterial diversity, we present a phylogeny based on comparisons of whole genomes in Fig. 26.15. As you examine this figure, remember that microbiologists continue to debate branching order on the bacterial tree.
26.4.2 What, if anything, is a bacterial species?
What do we mean when we talk about bacterial species? As we saw in Chapter 22, Ernst Mayr defined species as “groups of interbreeding natural populations that are reproductively isolated from other such groups.” This widely accepted definition is called the biological species concept (Chapter 22), but a moment’s reflection will show that it can’t be applied to the most abundant organisms on Earth—Bacteria and Archaea. In the absence of sexual reproduction, how do we think about species in the prokaryotic domains?
Some microbiologists argue that we should abandon the concept of species when talking about bacteria, especially since distantly related bacteria can exchange genes. Nonetheless, nature is full of microbial populations with well-defined features of form and function.
In the age of molecular sequence comparisons, bacterial species have sometimes been defined operationally: Two populations belong to the same species if the gene sequences of their small-subunit rRNA are more than 97% identical. It would be useful, however, to conceive of bacterial species in terms of population-level processes, much as Ernst Mayr did for eukaryotic organisms. In plants, animals, and single-celled eukaryotes, interbreeding ensures that populations share the same pool of genes, providing a counterforce to mutations and local selection pressures that promote divergence.
A similar process exists in bacteria. In early research on the population genetics of bacteria, biologists observed that genetic diversity in laboratory cultures gradually increases through time and then rapidly decreases with the emergence of a successful variant that outcompetes the rest. The episodic loss of diversity is called periodic selection. Some biologists argue that this process provides a means of recognizing bacterial species: Populations subject to the same episodes of periodic selection belong to a single species. This is a useful way to define bacterial species, but other definitions are possible, and as yet there is no consensus among microbiologists.
26-15
26.4.3 Proteobacteria are the most diverse bacteria.
Proteobacteria are far and away the most diverse of all bacterial groups (Fig. 26.15). Defined largely by similarities in gene sequences of rRNA, Proteobacteria include many of the organisms that populate our expanded carbon cycle and other biogeochemical cycles. For example, this phylum includes anoxygenic photosynthetic bacteria and chemoautotrophs that oxidize NH3, H2S, and Fe2+, as well as bacteria able to respire using SO42−, NO3−, or Fe3+.
Many Proteobacteria have evolved intimate ecological relationships with eukaryotic organisms. Some of these are mutually beneficial, such as the nitrogen-fixing bacteria in soybean root nodules. Other Proteobacteria, however, are our worst pathogens: the rickettsias that cause typhus and spotted fever, vibrios that trigger cholera, salmonellas responsible for typhoid fever and food poisoning, and Pseudomonas aeruginosa strains that infect burn victims and others whose resistance is reduced. Escherichia coli, famous as a laboratory model organism (see Fig. 26.4b), is usually a harmless presence in our intestinal tract, but pathogenic strains can cause serious diarrhea and even death.
26.4.4 The gram-positive bacteria include organisms that cause and cure disease.
In 1884, the Danish scientist Hans Christian Gram developed a diagnostic dye that, after washing, is retained by bacteria with thick peptidoglycan walls, but not by those with thin walls. Molecular sequence comparisons confirm that most gram-positive bacteria (those that retain the dye) form a well-defined branch of the bacterial tree (Fig. 26.15). One species in this group, Bacillus subtilis, is well known to biologists because it has been the focus of many studies about basic molecular function in bacteria. All of us harbor Streptococcus species in our mouths, and many of us have suffered from strep throat, produced by streptococcal pathogens. More serious infections, including some forms of meningitis, are also associated with streptococci. Staphylococcus bacteria are commonly found on human skin, but only occasionally do we suffer from maladies such as staph infection, toxic shock syndrome, or food poisoning induced by staphylococcal bacteria.
In contrast to these pathogens, gram-positive bacteria called streptomycetes have proved invaluable to medicine because of a remarkable property. More than half of the species in this group secrete compounds that kill other bacteria and fungi in their vicinity. Streptomycetes have provided us with tetracycline, streptomycin, erythromycin, and numerous other antibiotics that combat infectious disease.
26.4.5 Photosynthesis is widely distributed on the bacterial tree.
As noted earlier, molecular studies confirm that all bacteria capable of oxygenic photosynthesis form a single branch, the cyanobacteria. Cyanobacteria can be found in environments that range from deserts to the open ocean. Their diverse species include unicellular rods and spheroidal cells, as well as multicellular balls and filaments (Fig. 26.16). Some filamentous cyanobacteria can even form several different cell types, including specialized cells for nitrogen fixation and resting cells that provide protection when the local environment does not favor growth.
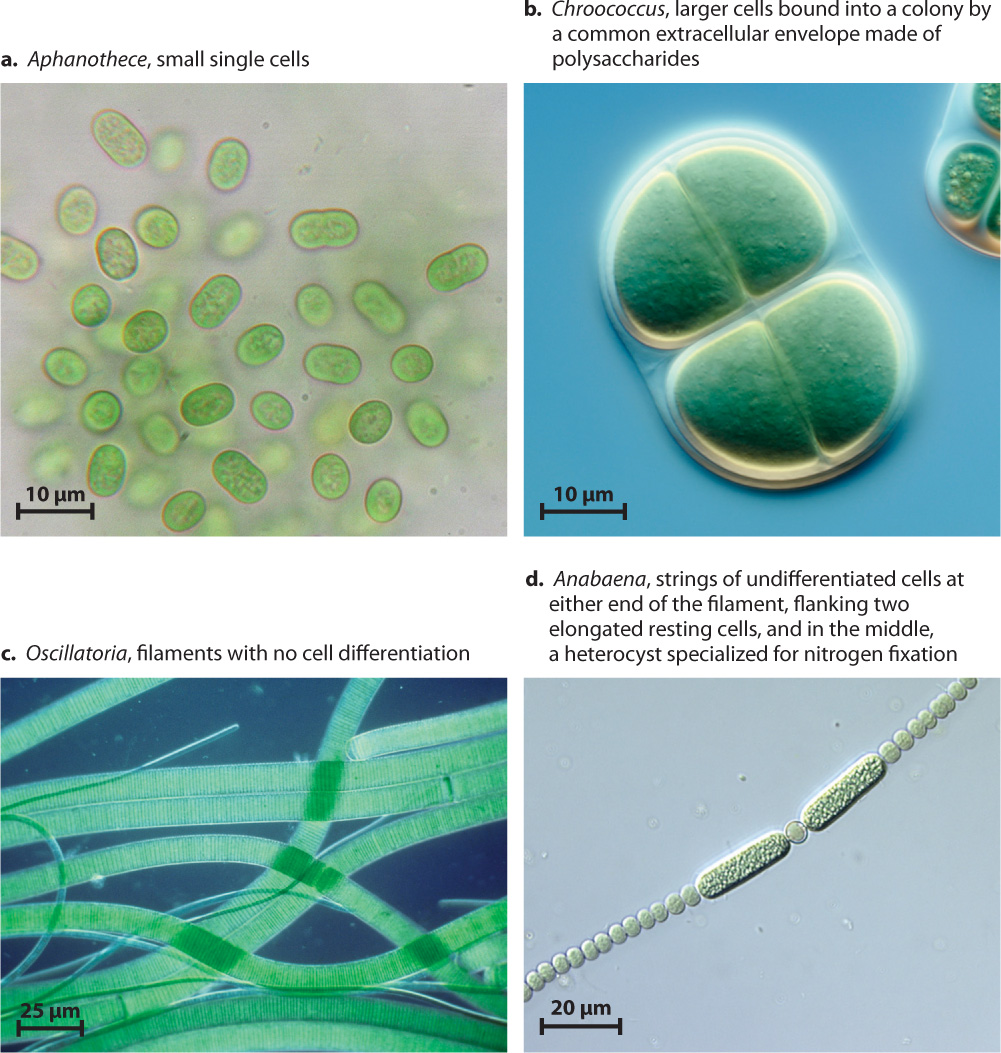
26-16
In contrast to the monophyly of cyanobacteria, molecular sequence comparisons show that anoxygenic photosynthesis is distributed widely on the bacterial tree (see Fig. 26.15). Think of the purple layer beneath the surface of many microbial mats (see Fig. 26.7). The vivid color comes from the light-sensitive pigments of the aptly named purple bacteria, found within the Proteobacteria. Purple bacteria are capable of photoautotrophic growth using bacteriochlorophyll and a single photosystem. Most, however, show evidence of metabolic diversity and can grow heterotrophically in the absence of light or appropriate electron donors. The green layer beneath the surface of some microbial mats reflects the pigments of another group of photosynthetic bacteria, called green sulfur bacteria because they commonly gain electrons from hydrogen sulfide and deposit elemental sulfur on their walls. Unlike most groups of photosynthetic bacteria, the green sulfur bacteria are highly intolerant of oxygen gas. Oddly, a species of green sulfur bacteria has been found in hydrothermal rift vents 2 km beneath the surface of the ocean. Light doesn’t penetrate to these depths, so if these organisms harvest light, it must come from incandescent lavas as they erupt.
Light-harvesting bacteria occur on several other branches of the bacterial tree—the photoheterotrophic heliobacteria, for example, and the green nonsulfur bacteria often found in freshwater hot springs. Despite a century of research, our knowledge of photosynthesis within the bacterial tree remains incomplete, as illustrated by the discovery in 2007 of unique light-harvesting bacteria in Yellowstone National Park. Genomic surveys of hot springs turned up evidence of a previously unknown bacteriochlorophyll-containing microorganism, and gene sequencing established that this organism belongs to the phylum Acidobacteria. There is no evidence for carbon fixation, however, suggesting that the Yellowstone bacterium is a photoheterotroph. To date, these unusual microorganisms have not been grown in pure culture, so as yet they are difficult to study.