9.5 The Proteome
Chapter 8 began with a discussion of the number of genes in the human genome and how that number (about 21,000) was much lower than the actual number of proteins in a human cell (more than 100,000). Now that you are familiar with how information encoded in DNA is transcribed into RNA and how RNA is translated into protein, it is a good time to revisit this matter and look more closely at the sources of protein diversification. First, let’s review a few old terms and add a new one that will be useful in this discussion. You already know that the genome is the entire set of genetic material in an organism. You will learn in Chapter 14 that the transcriptome is the complete set of coding and noncoding transcripts in an organism, organ, tissue, or cell. Another term is the proteome, which was briefly introduced in Chapter 8 but is defined here as the complete set of proteins in an organism, organ, tissue, or cell. In the remainder of this chapter, you will see how the proteome is enriched by two cellular processes: the alternative splicing of pre-
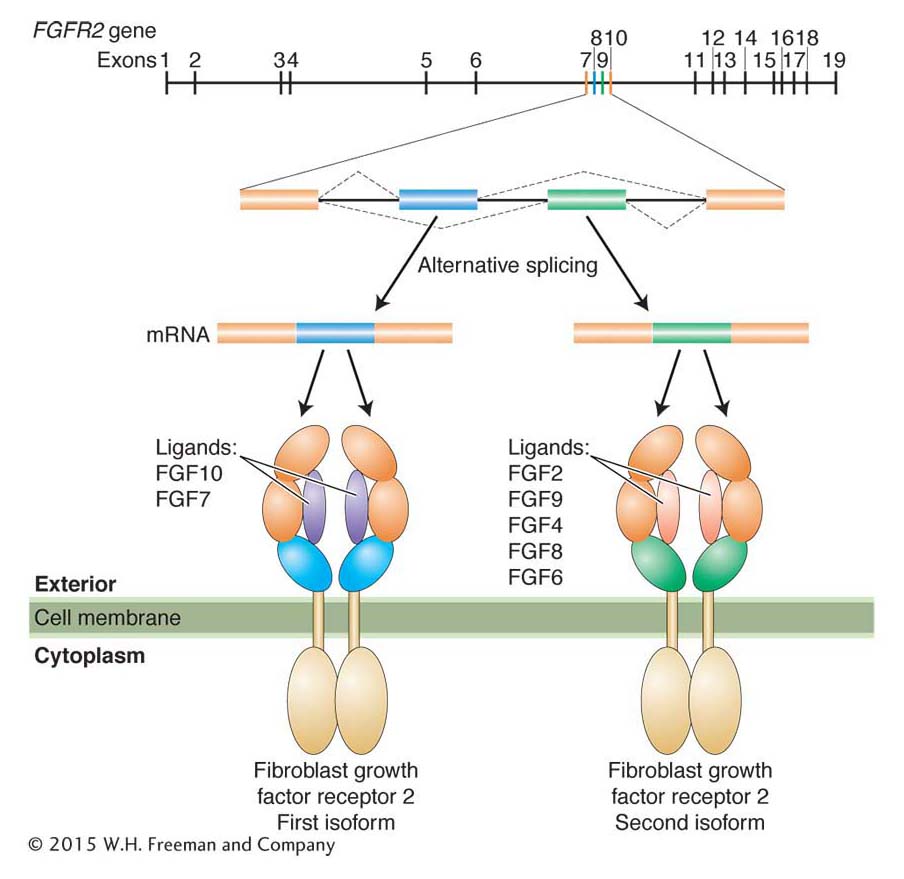
Alternative splicing generates protein isoforms
As you recall from Chapter 8, alternative splicing of pre-
340
Posttranslational events
When released from the ribosome, most newly synthesized proteins are unable to function. As you will see in this section and in subsequent chapters of this book, DNA sequence is only part of the story of how organisms function. In this case, all newly synthesized proteins need to fold up correctly and the amino acids of some proteins need to be chemically modified. Because some protein folding and modification take place after protein synthesis, they are called posttranslational events.
Protein folding inside the cell The most important posttranslational event is the folding of the nascent (newly synthesized) protein into its correct three-
341
The answer seems to be that nascent proteins are folded correctly with the help of chaperones—
Posttranslational modification of amino acid side chains As already stated, proteins are polymers of amino acids made from any of the 20 different types. However, biochemical analysis of many proteins reveals that a variety of molecules can be covalently attached to amino acid side chains. More than 300 modifications of amino acid side chains are possible after translation. Two of the more commonly encountered posttranslational modifications—
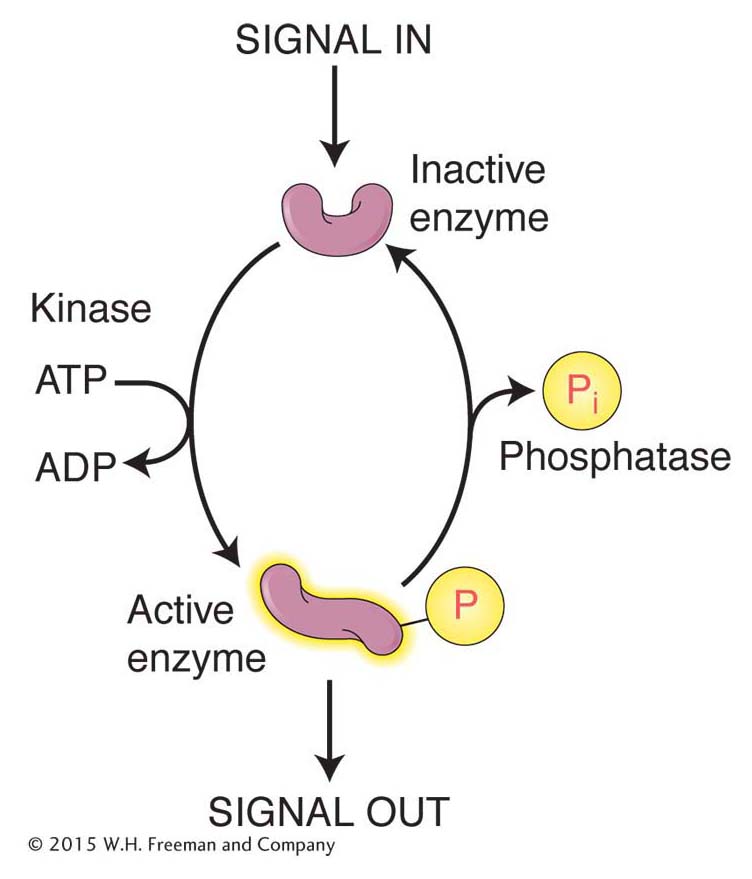
Phosphorylation Enzymes called kinases attach phosphate groups to the hydroxyl groups of the amino acids serine, threonine, and tyrosine, whereas enzymes called phosphatases remove these phosphate groups. Because phosphate groups are negatively charged, their addition to a protein usually changes protein conformation. The addition and removal of phosphate groups serves as a reversible switch to control a variety of cellular events, including enzyme activity, proteinprotein interactions, and protein–
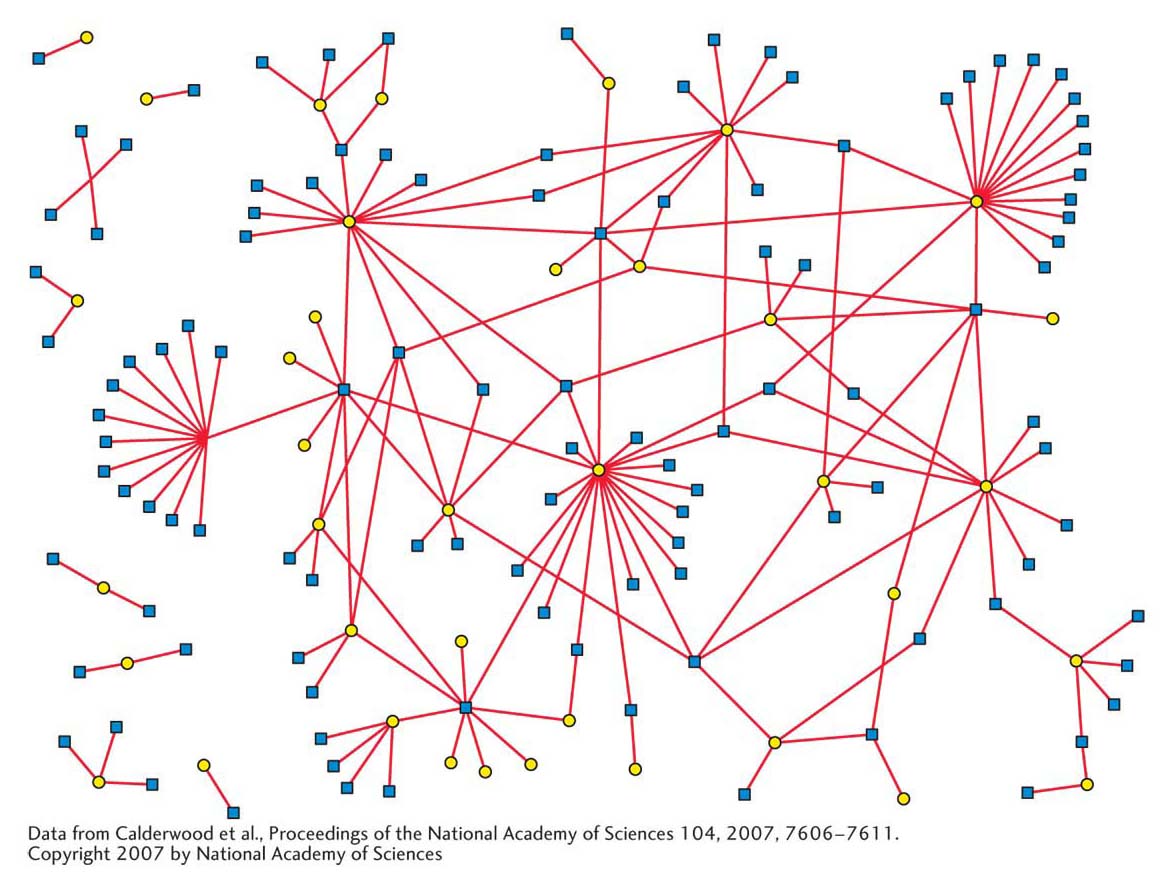
One measure of the importance of protein phosphorylation is the number of genes encoding kinase activity in the genome. Even a simple organism such as yeast has hundreds of kinase genes, whereas the mustard plant Arabidopsis thaliana has more than 1000. Another measure of the significance of protein phosphorylation is that most of the numerous protein–
Recent analyses of the protein–
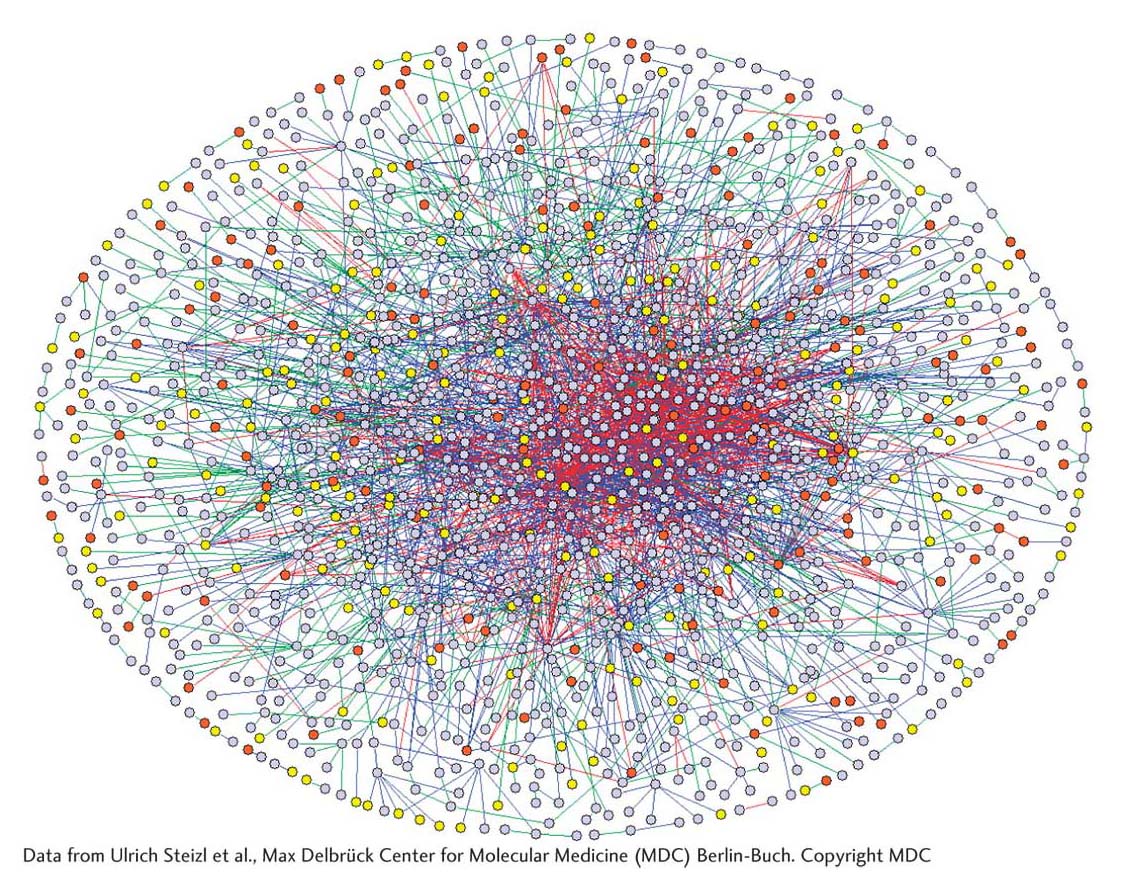
What is the biological significance of these interactions? In this chapter and preceding ones, you have seen that protein–
Ubiquitination Surprisingly, one of the most common posttranslational modifications is not a subtle one like the addition of a phosphate group. Instead, this modification targets the protein for degradation by a biological machine and protease called the 26S proteasome (Figure 9-23). The modification targeting a protein for degradation is the addition of chains of multiple copies of a protein called ubiquitin to the ε-amine of lysine residues (called ubiquitination). Ubiquitin contains 76 amino acids and is found only in eukaryotes, where it is highly conserved in plants and animals. Two broad classes of proteins are targeted for destruction by ubiquitination: short-
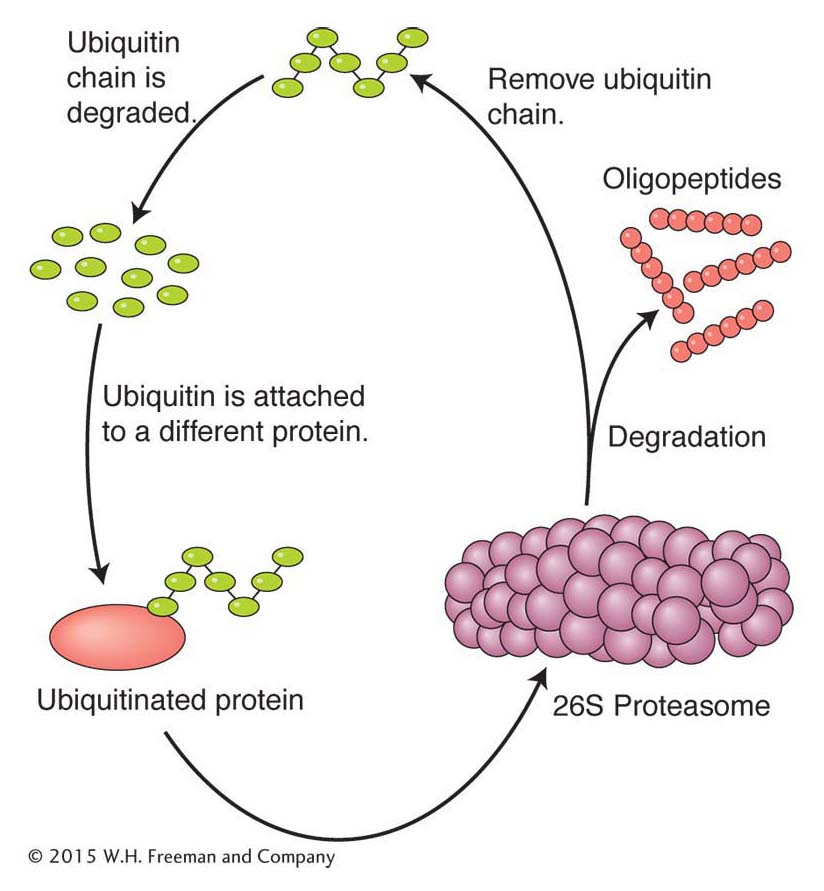
342
343
Protein targeting In eukaryotes, all proteins are synthesized on ribosomes in the cytoplasm. However, some of these proteins end up in the nucleus, others in the mitochondria, and still others anchored in the membrane or secreted from the cell. How do these proteins “know” where they are supposed to go? The answer to this seemingly complex problem is actually quite simple: a newly synthesized protein contains a short sequence that targets the protein to the correct place or cellular compartment. For example, a newly synthesized membrane protein or a protein destined for an organelle has a short leader peptide, called a signal sequence, at its amino-
Proteins destined for the nucleus include the RNA and DNA polymerases and transcription factors discussed in Chapters 7 and 8. Amino acid sequences embedded in the interiors of such nucleusbound proteins are necessary for transport from the cytoplasm into the nucleus. These nuclear localization sequences (NLSs) are recognized by cytoplasmic receptor proteins that transport newly synthesized proteins through nuclear pores—
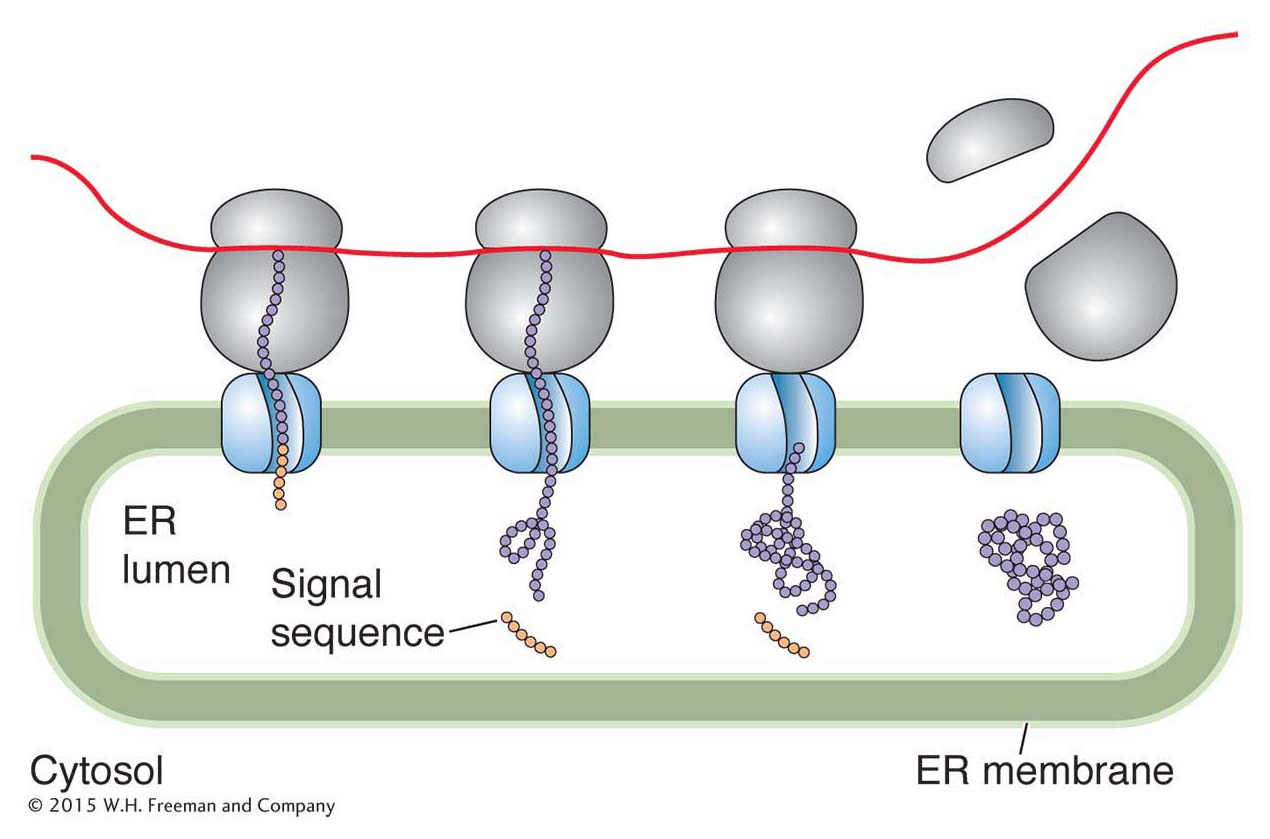
Why are signal sequences cleaved during targeting, whereas an NLS, located in a protein’s interior, remains after the protein moves into the nucleus? One explanation might be that, in the nuclear disintegration that accompanies mitosis (see Chapter 2), proteins localized to the nucleus may find themselves in the cytoplasm. Because such a protein contains an NLS, it can relocate to the nucleus of a daughter cell that results from mitosis.
KEY CONCEPT
Most eukaryotic proteins are inactive unless modified after translation. Some posttranslational events, such as phosphorylation or ubiquitination, modify amino acid side groups, thus promoting protein activation or degradation, respectively. Other posttranslational mechanisms recognize amino acid signatures in a protein sequence and target those proteins to places where their activity is required inside or outside the cell.344