10.2 Generating Recombinant DNA Molecules
Recombinant DNA molecules usually contain a DNA fragment inserted into a bacterial vector. In this section, you will see that there are many types of recombinant DNA molecules that can be constructed from a variety of donor DNAs and vectors. We begin by discussing sources of donor DNA:
355
If the experimenter wants a collection of inserts that represents the entire genome of an organism, the genomic DNA can be cut up before cloning.
Alternatively, if the goal is to isolate a single gene, the polymerase chain reaction can be used to amplify selected regions of DNA in vitro.
Finally, if the researcher desires only the coding sequences of genes, without introns, DNA copies of the mRNA products, called cDNA, can be synthesized and inserted into a vector.
Genomic DNA can be cut up before cloning
Genomic DNA is obtained directly from the chromosomes of the organism under study, usually by grinding up fresh tissue and purifying the DNA. Chromosomal DNA can be used as the starting point for both in vivo and PCR methods to isolate genes. For the in vivo method, genomic DNA needs to be cut up before cloning is possible. As described later in this section, genomic DNA does not have to be cut up to perform PCR because the specific short primers that anneal to it identify the start site for DNA polymerase that directs the replication of the intervening DNA.
The long chromosome-
Another key property of some restriction enzymes is that many create “sticky ends” in the fragments. Let’s look at an example. The restriction enzyme EcoRI (from E. coli) recognizes the following sequence of six nucleotide pairs in the DNA of any organism:
5′-GAATTC-
3′-CTTAAG-
This type of segment is called a DNA palindrome, which means that both strands have the same nucleotide sequence but in antiparallel orientation (reading 5′ to 3′ produces the same sequence on either strand). Different restriction enzymes cut at different palindromic sequences. Sometimes the cuts are in the same position on each of the two antiparallel strands, leaving blunt ends. However, the most useful restriction enzymes make cuts that are offset, or staggered. The enzyme EcoRI makes cuts only between the G and the A nucleotides on each strand of the palindrome:
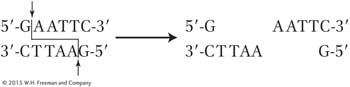
These staggered cuts leave a pair of ends that each have an identical four base-
356
Digesting human genomic DNA with EcoRI generates approximately 500,000 fragments. You will see later in this section how scientists sift through all of these fragments to find the needle in the haystack—
KEY CONCEPT
Genomic DNA can be used directly for cloning genes. As a first step, restriction enzymes cut DNA into fragments of manageable size, and many of them generate single-The polymerase chain reaction amplifies selected regions of DNA in vitro
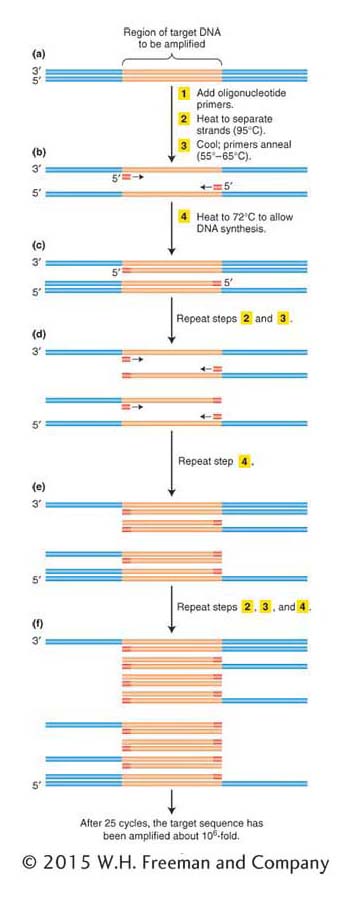
ANIMATED ART: Polymerase chain reaction
If we endeavored to clone the human insulin gene today, armed with the human genome sequence, knowing the gene and flanking sequences would allow us to use a more direct method. Today, we can simply amplify the gene in vitro using the polymerase chain reaction (PCR). The basic strategy of PCR is outlined in Figure 10-3. The process uses multiple copies of a pair of short chemically synthesized DNA primers, approximately 20 bases long, designed so that each primer will bind to one end of the gene or region to be amplified. The two primers bind to opposite DNA strands surrounding the target sequence, with their 3′ ends pointing toward each other. DNA polymerases add bases to the 3′ ends of these primers and copy the target sequence. Repeating the polymerization process produces an exponentially growing number of double-
We start with a solution containing the DNA template, the primers, the four deoxyribonucleotide triphosphates (required for DNA synthesis; see Figure 7-
After the replication of the segment between the two primers is completed, the two new duplexes are again heat-
357
PCR is a powerful technique that is routinely used to isolate specific genes or DNA fragments when there is prior knowledge of the sequence to be amplified. In fact, if the sequences corresponding to the primers are each present only once in the genome and are sufficiently close together, the only DNA segment that can be amplified is the one between the two primers. PCR is a very sensitive technique with numerous applications in biology. It can amplify target sequences that are present in extremely low copy numbers in a sample as long as primers specific to this rare sequence are used. For example, crime investigators can amplify segments of human DNA from the few follicle cells surrounding a single pulled-
It would not be an overstatement to say that PCR has revolutionized the study of many fields of biology where DNA analysis is required. In recognition of its importance to science, Kary Mullis was awarded the Nobel Prize in Chemistry in 1993 for developing the first viable PCR protocol.
358
KEY CONCEPT
The polymerase chain reaction uses specially designed primers for direct isolation and amplification of specific regions of DNA in a test tube.DNA copies of mRNA can be synthesized
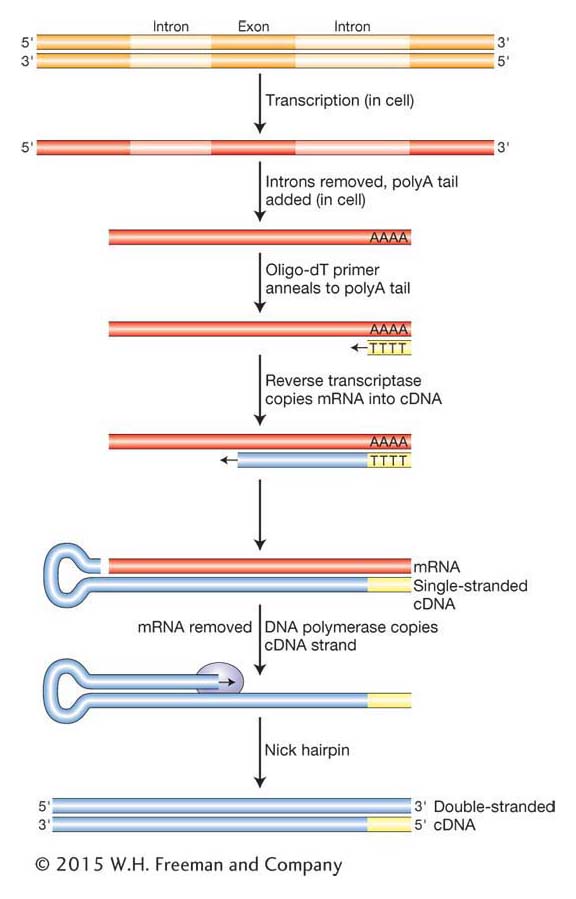
As we have seen in Chapter 8, eukaryotic genes often contain one or more introns that disrupt the coding regions. Further, as we will see in Chapters 14 and 15, protein-
Complementary DNA (cDNA) is a DNA version of an mRNA molecule. Researchers use cDNA rather than mRNA itself because RNAs are inherently less stable than DNA. Moreover, RNA cannot be manipulated by the enzymes available for DNA cloning, and techniques for routinely amplifying and purifying individual RNA molecules do not exist. The cDNA is made from mRNA with the use of a special enzyme called reverse transcriptase, originally isolated from retroviruses (see Chapter 15). Retroviruses have RNA genomes that are copied into DNA that inserts into the host chromosome. Can you think of why it is called reverse transcriptase? To make cDNA, a researcher begins by purifying mRNA from a tissue that produces a large amount of the desired protein. Insulin is produced in the β-islet cells of the pancreas, so we would use that organ as our source for insulin mRNA. Next, the purified mRNA is added to a test tube containing reverse transcriptase, the four dNTPs, and a short primer of polymerized dTTP residues (called an oligo-
KEY CONCEPT
mRNA is often a preferable starting point in the isolation of a gene. Enzymatic conversion of mRNA into cDNA allows for the isolation of a gene copy without introns.Attaching donor and vector DNA
As described above, we have several options for obtaining the human insulin gene from the genome or from purified mRNA. These methods produce genomic DNA fragments, PCR products, or double-
359
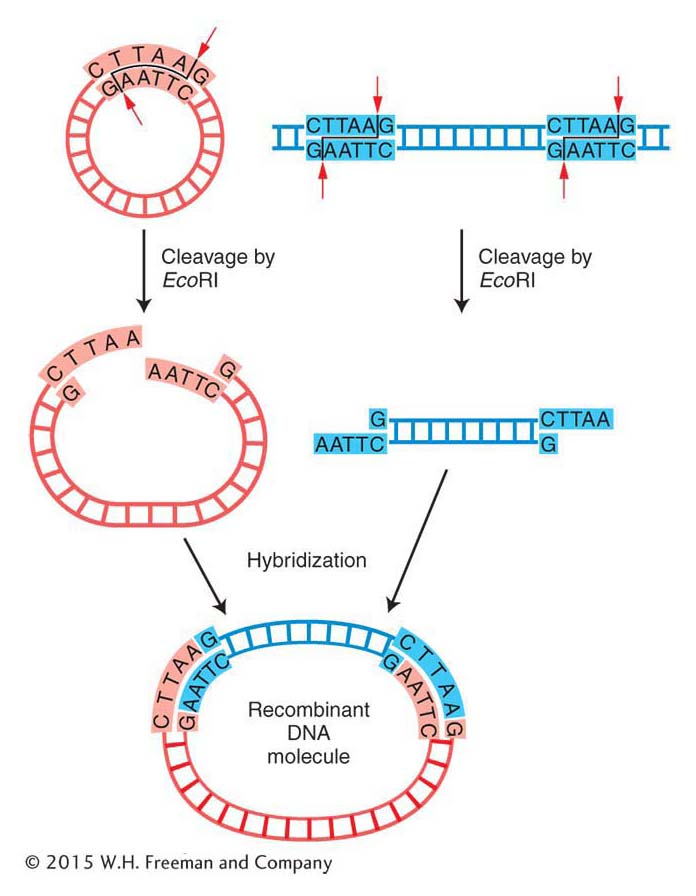
Cloning DNA fragments with sticky ends Recall that the original scientists who isolated the human insulin gene did not know the gene sequence, and so they needed to create alibrary of human genome DNA fragments from which to isolate the specific gene. To make recombinant DNA molecules containing donor genomic DNA fragments, both donor and vector DNAs are digested by a restriction enzyme that produces the same complementary sticky ends (see Figure 10-2). The resulting fragments are then mixed to allow the sticky ends of vector and donor DNA to hybridize with each other and form recombinant molecules. Figure 10-5a shows a bacterial plasmid DNA that carries a single EcoRI restriction site, so digestion with the restriction enzyme EcoRI converts the circular DNA into a single linear molecule with sticky ends. Donor DNA from any other source, such as human DNA, also is treated with the EcoRI enzyme to produce a population of fragments carrying the same sticky ends. When the two populations are mixed under the proper physiological conditions, DNA fragments from the two sources can hybridize because double helices form between their sticky ends (Figure 10-5b). In any cloning reaction, there are many linearized plasmid molecules in the solution, as well as many different EcoRI fragments of donor DNA, a tiny fraction of which will have the target DNA. Therefore, a diverse array of plasmids recombined with different donor fragments will be produced. At this stage, the hybridized molecules do not have covalently joined sugar-
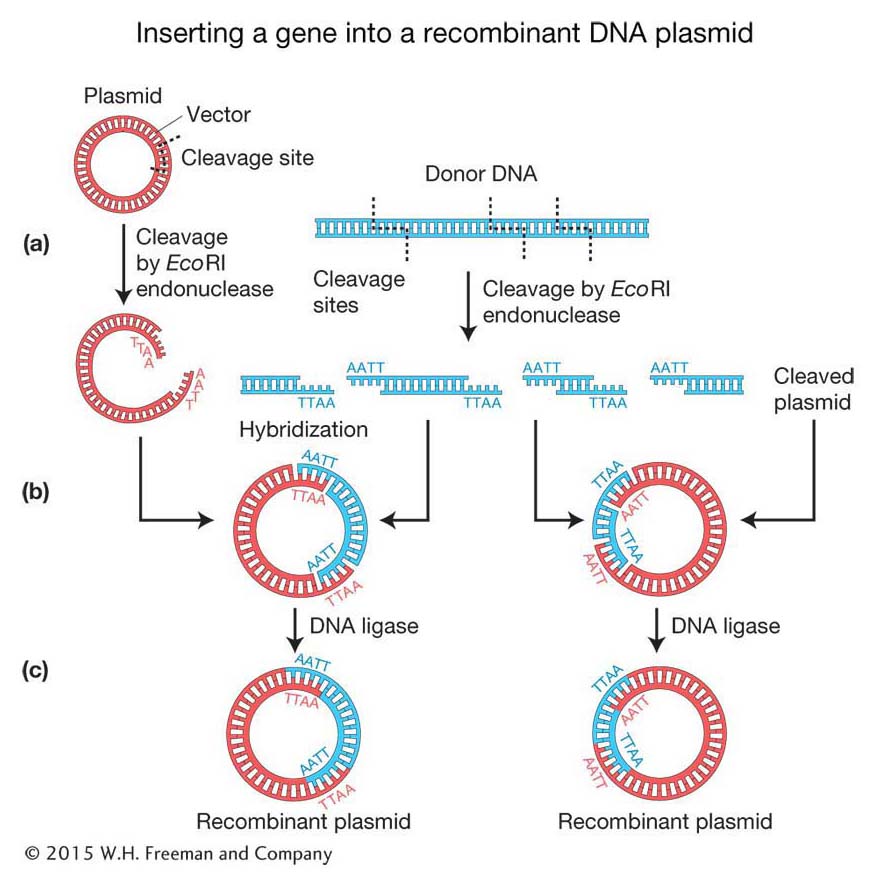
360
Cloning DNA fragments with blunt ends Knowing the human insulin gene sequence helps us zero in on the gene, but it adds a small complication in the cloning reaction. Some restriction enzymes produce blunt ends rather than staggered cuts. In addition, cDNA and the DNA fragments that arise from PCR have blunt or near-
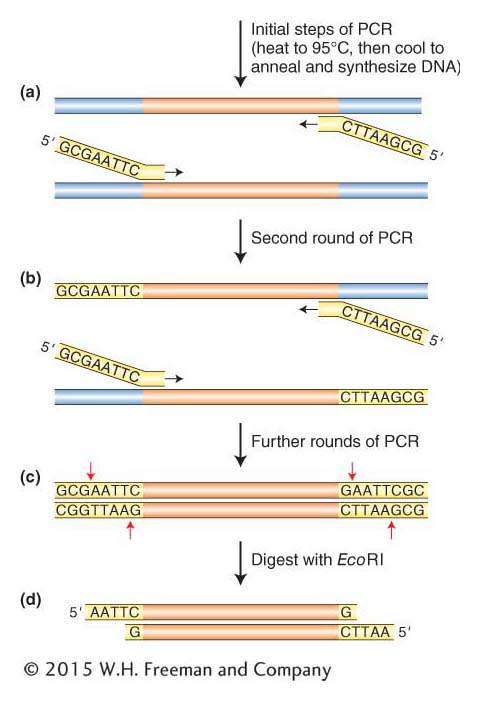
361
Another method adds sticky ends to any double-
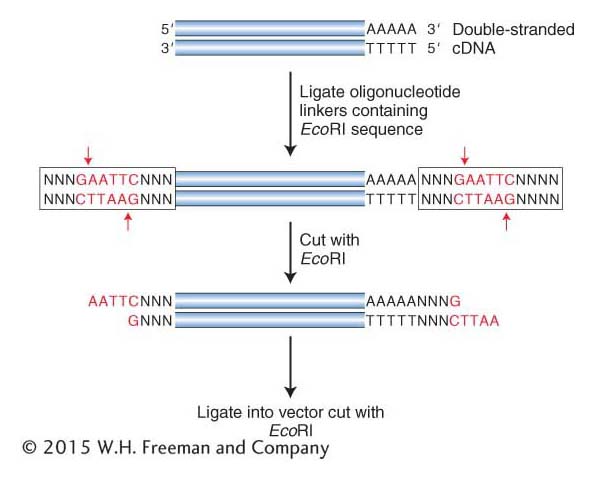
362
KEY CONCEPT
Donor and vector DNAs with the same sticky ends can be joined efficiently and ligated. Alternatively, donor DNA that is the product of PCR or cDNA synthesis requires the addition of sticky ends prior to insertion into a vector.Amplification of donor DNA inside a bacterial cell
Amplification of the recombinant DNA molecules takes advantage of prokaryotic genetic processes such as bacterial transformation, plasmid replication, and bacteriophage growth, all discussed in Chapter 5. Figure 10-8 illustrates the cloning of a donor DNA segment. A single recombinant vector enters a bacterial cell and is amplified by the same machinery that replicates the bacterial chromosome. One basic requirement is the presence of an origin of DNA replication recognized by the host replication proteins (as described in Chapter 7). There are soon many copies of each vector in each bacterial cell. Hence, after amplification, a colony of bacteria will typically contain billions of copies of the single-
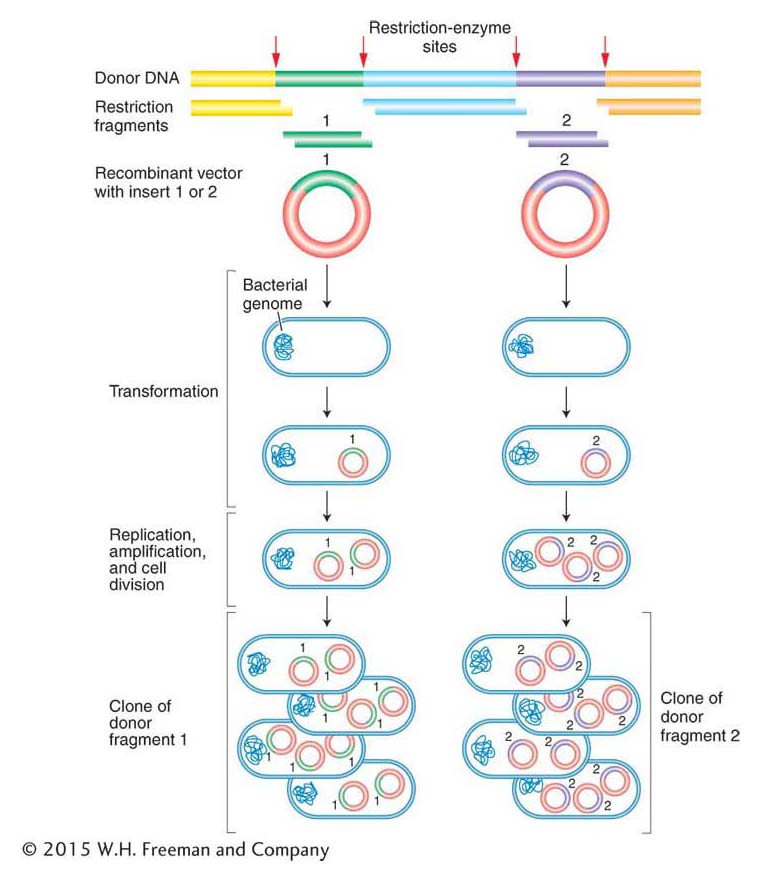
ANIMATED ART: Finding specific cloned genes by functional complementation: making a library of wild-
363
The amplification of donor DNA inside a bacterial cell entails the following steps:
Choosing a cloning vector and introducing the insert (see the preceding section for a discussion of the latter)
Introducing the recombinant DNA molecule inside a bacterial cell
Recovering the amplified recombinant molecules
Choice of cloning vectors Vectors must be small molecules for convenient manipulation, but they may vary in many ways to suit the goal of the experiment. Some vectors need to be capable of prolific replication in a living cell in order to amplify the inserted donor fragment. In contrast, others are designed to be present in only a single copy to maintain the integrity of the inserted DNA (see below). All vectors must have convenient restriction sites at which the DNA to be cloned may be inserted (called a polylinker or multiple cloning site). Ideally, the restriction site should be present only once in the vector because then restriction fragments of donor DNA will insert only at that one location in the vector. Having a way to identify and recover the desired recombinant molecule quickly also is important. Numerous cloning vectors that meet a wide range of experimental needs are in current use. Some general classes of cloning vectors follow.
Plasmid vectors As described earlier, bacterial plasmids are small circular DNA molecules that commonly replicate their DNA independent of the bacterial chromosome. The plasmids routinely used as vectors carry a gene for drug resistance and a gene to distinguish plasmids with and without DNA inserts. These drug-
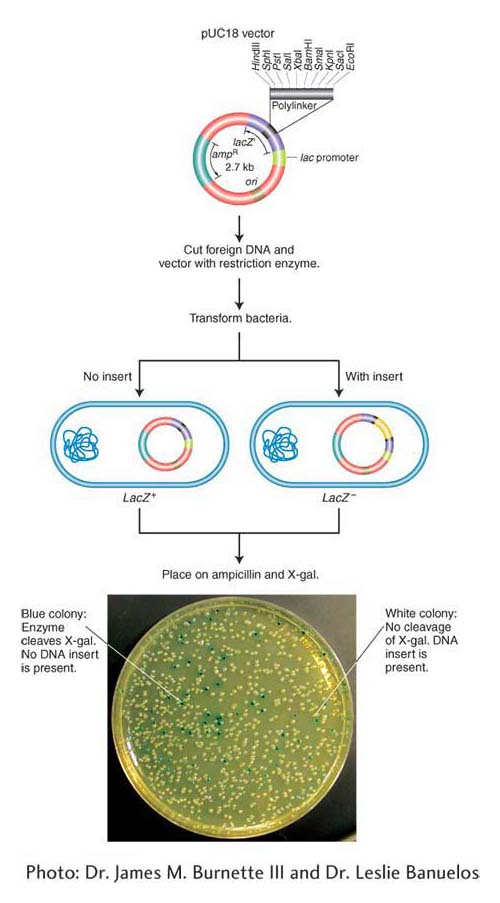
Bacteriophage vectors A bacteriophage vector harbors DNA as an insert “packaged” inside the phage particle. Different classes of bacteriophage vectors can carry different sizes of donor DNA insert. Bacteriophage λ (lambda; discussed in Chapters 5 and 11) is an effective cloning vector for double-
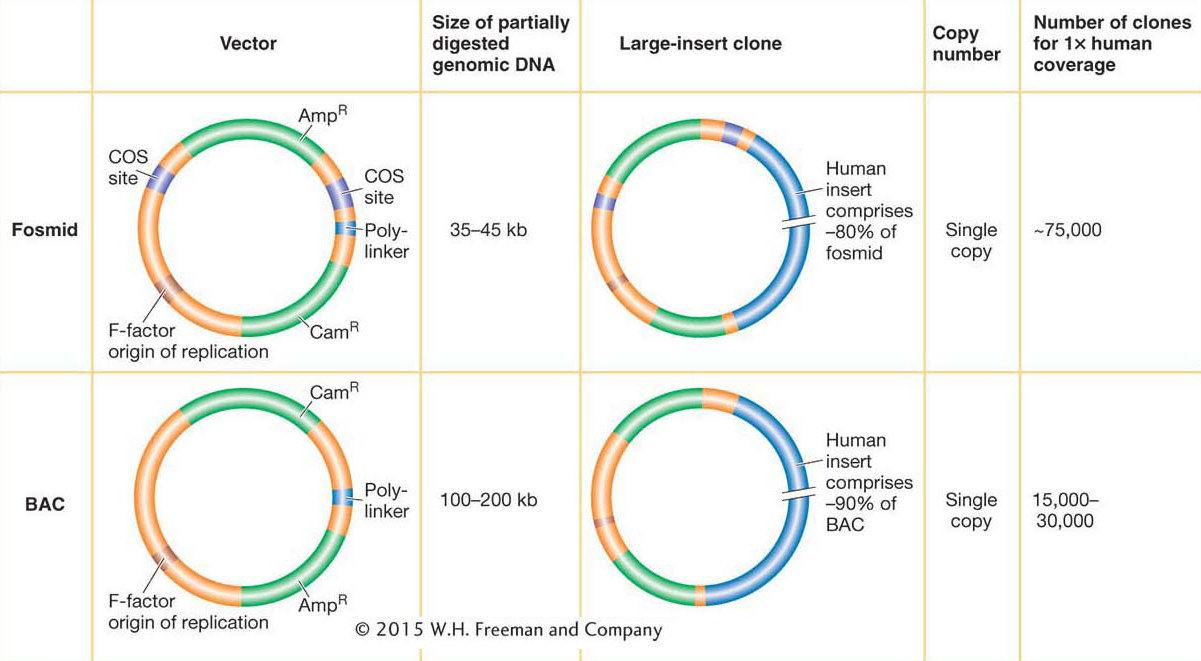
Vectors for larger DNA inserts The standard plasmid and phage λ vector just described can accept donor DNA of sizes as large as 10 to 15 kb. However, many experiments require inserts well in excess of this upper limit. To meet these needs, special vectors that require more sophisticated methods for transferring the DNA into the host cell have been engineered. In each case, the DNAs replicate as large plasmids after they have been delivered into the bacterium.
364
Fosmids are vectors that can carry 35-
365
The most popular vector for cloning very large DNA inserts in bacteria is the bacterial artificial chromosome (BAC). Derived from the F plasmid, it can carry inserts ranging from 100 to 200 kb, although the vector itself is only ~7 kb (see Figure 10-10). The DNA to be cloned is inserted into the plasmid, and this large circular recombinant DNA is introduced into the bacterium. BACs were the “workhorse” vectors for the extensive cloning required by large-
KEY CONCEPT
The genetic engineer’s toolkit contains a variety of cloning vectors that accept inserts of small sizes for plasmids, to medium sizes for bacteriophage, to large sizes for fosmids and BACs.Entry of recombinant molecules into the bacterial cell Three methods are used to introduce recombinant DNA molecules into bacterial cells: transformation, transduction, and infection (Figure 10-11; see Sections 5.3 and 5.4).
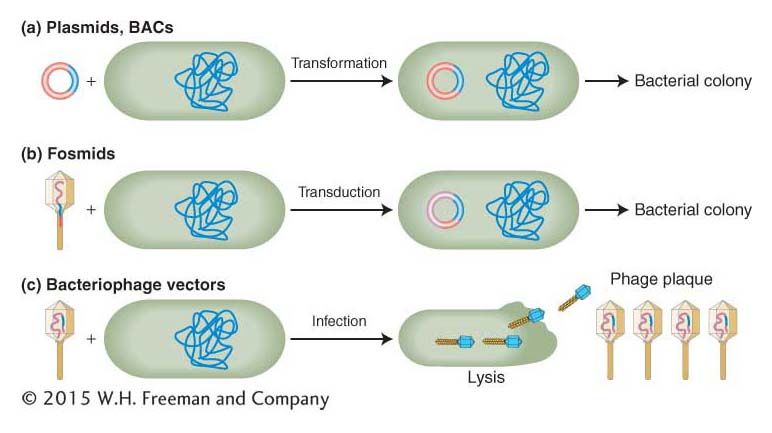
In transformation, bacteria are bathed in a solution containing the recombinant DNA molecule. Because bacterial cells used in research cannot take up DNA molecules as large as recombinant plasmids, they must be made competent (that is, able to take up the DNA from the surrounding media) by either incubation in a calcium solution or exposure to a high-
voltage electrical pulse (electroporation). After entering a competent cell through membrane pores, the recombinant molecule becomes a plasmid chromosome (Figure 10-11a). Electroporation is the method of choice for introducing especially large DNAs such as BACs into bacterial cells.
366
In transduction, the recombinant molecule is combined with phage head and tail proteins to produce a virus that contains largely non-
viral DNA. These engineered phages are then mixed with bacteria and they inject their DNA cargo into the bacterial cells, but new phages cannot form because they do not carry the viral genes necessary for phage replication. Fosmids are introduced into cells by transduction (Figure 10-11b). In contrast to transduction, which produces plasmids and bacterial colonies but not new viruses, infection produces recombinant phage particles (Figure 10-11c). Through repeated rounds of re-
infection, a plaque full of phage particles forms from each initial bacterium that was infected. Each phage particle in a plaque contains not only the recombinant DNA but also viral genes needed to create new infective phage particles.
Recovery of amplified recombinant molecules The recombinant DNA packaged into phage particles is easily obtained by collecting phage lysate and isolating the DNA that they contain. To obtain the recombinant DNA packaged in plasmids, fosmids, or BACs, the bacteria are chemically or mechanically broken apart. The recombinant DNA plasmid is separated from the much larger main bacterial chromosome by centrifugation, electrophoresis, or other selective techniques that distinguish the chromosome from the plasmid by size or shape.
KEY CONCEPT
Gene cloning is carried out through the introduction of single recombinant vectors into recipient bacterial cells, followed by the amplification of these molecules as either plasmid chromosomes or phages.Making genomic and cDNA libraries
We have seen how to make and amplify individual recombinant DNA molecules such as our human insulin cDNA. Consider the task in 1982, when the human insulin gene had to be identified from a library of human genome fragments. To ensure that we have cloned the DNA segment of interest, we have to make large collections of DNA segments that are all-
367
Similarly, representative collections of cDNA inserts require tens or hundreds of thousands of independent cDNA clones; these collections are cDNA libraries and represent only the protein-
Whether we choose to construct a genomic DNA library or a cDNA library depends on the situation. If we are seeking a specific gene that is active in a specific type of tissue in a plant or animal, then it makes sense to construct a cDNA library from a sample of that tissue. For example, suppose we want to identify cDNAs corresponding to insulin mRNAs. The β-islet cells of the pancreas are the most abundant source of insulin, and so mRNAs from pancreas cells are the appropriate source for a cDNA library because these mRNAs should be enriched for the gene in question. A cDNA library represents a subset of the transcribed regions of the genome; so it will inevitably be smaller than a complete genomic library. Although genomic libraries are bigger, they do have the benefit of containing genes in their native form, including introns and untranscribed regulatory sequences. A genomic library is necessary at some stage as a prelude to cloning an entire gene or an entire genome.