10.3 Using Molecular Probes to Find and Analyze a Specific Clone of Interest
The production of a library as just described is sometimes referred to as “shotgun” cloning because the experimenter clones a large sample of fragments and hopes that one of the clones will contain a “hit”—the desired gene. The task then is to find that particular clone, considered next.
Finding specific clones by using probes
A library might contain as many as hundreds of thousands of cloned DNA fragments. This huge collection of fragments must be screened to find the recombinant DNA molecule containing the gene of interest to a researcher. Such screening is accomplished by using a specific probe that will find and mark only the desired clone. There are two types of probes: (1) those that recognize a specific nucleic acid sequence and (2) those that recognize a specific protein.
368
Probes for finding DNA Probing for DNA makes use of the power of base complementarity. Two single-
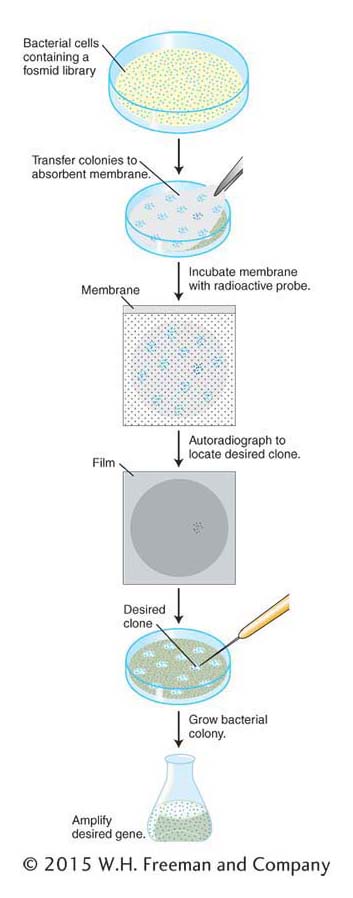
The identification of a specific clone in a library is a multistep procedure. In Figure 10-12, these steps are shown for a library cloned into a fosmid vector. The steps are similar for libraries of plasmids or BACs. For libraries of phages, plaques are screened rather than colonies. First, colonies of the library on a petri dish are transferred to an absorbent membrane by simply laying the membrane on the surface of the medium. The membrane is peeled off, colonies clinging to the surface are lysed in place on the membrane, and the DNA is simultaneously denatured so that it is single-
Where does the DNA to make a probe come from? The DNA can come from one of several sources.
One can use a homologous gene or a cDNA from a related organism. This method depends on the fact that organisms descended from a recent common ancestor will have similar DNA sequences. Even though the probe DNA and the DNA of the desired clone might not be identical, they are often similar enough to promote hybridization.
369
One can use the protein product of the gene of interest. If part or all of the protein sequence is known, one can back-
translate, by using the table of the genetic code in reverse (from amino acid to codon), to deduce the DNA sequences that may have encoded it. A synthetic DNA probe that matches that sequence is then designed. Recall, however, that the genetic code is degenerate— that is, most amino acids are encoded by multiple codons. Thus, several possible DNA sequences could in theory encode the protein in question, but only one of these DNA sequences is present in the gene that actually encodes the protein. To get around this problem, a short stretch of amino acids with minimal degeneracy is selected. A mixed set of probes is then designed containing all possible DNA sequences that can encode this amino acid sequence. This “cocktail” of oligonucleotides is used as a probe, and a correct or very similar strand within this cocktail will hybridize with the gene of interest. Oligonucleotides of about 20 nucleotides in length embody enough specificity to hybridize to one unique complementary DNA sequence in the library.
Probes for finding proteins If the protein product of a gene is known and isolated in pure form, then this protein can be used to detect the clone of the corresponding gene in a library. The process, described in Figure 10-13, requires two components. First, it requires an expression library, made by using expression vectors that will direct the host cell to produce the protein. To make the library, cDNA is inserted into the vector in the correct triplet reading frame with a bacterial promoter, and cells containing the vector and its insert produce a translation of the cDNA insert. Second, the process requires an antibody that binds to the specific protein product of the gene of interest. (An antibody is a protein made by an animal’s immune system that binds with high affinity to a given molecule.) The antibody is used to screen the expression library for that protein. A membrane is lain over the surface of the medium and removed so that some of the cells of each colony are now attached to the membrane at locations that correspond to their positions on the original petri dish (see Figure 10-13). The imprinted membrane is then dried and bathed in a solution of the antibody, which will bind to the imprint of any colony that contains the fusion protein of interest. Positive clones are revealed by a labeled secondary antibody that binds to the first antibody. By detecting the correct protein, the antibody identifies the clone containing the gene that must have synthesized that protein and therefore contains the desired cDNA.
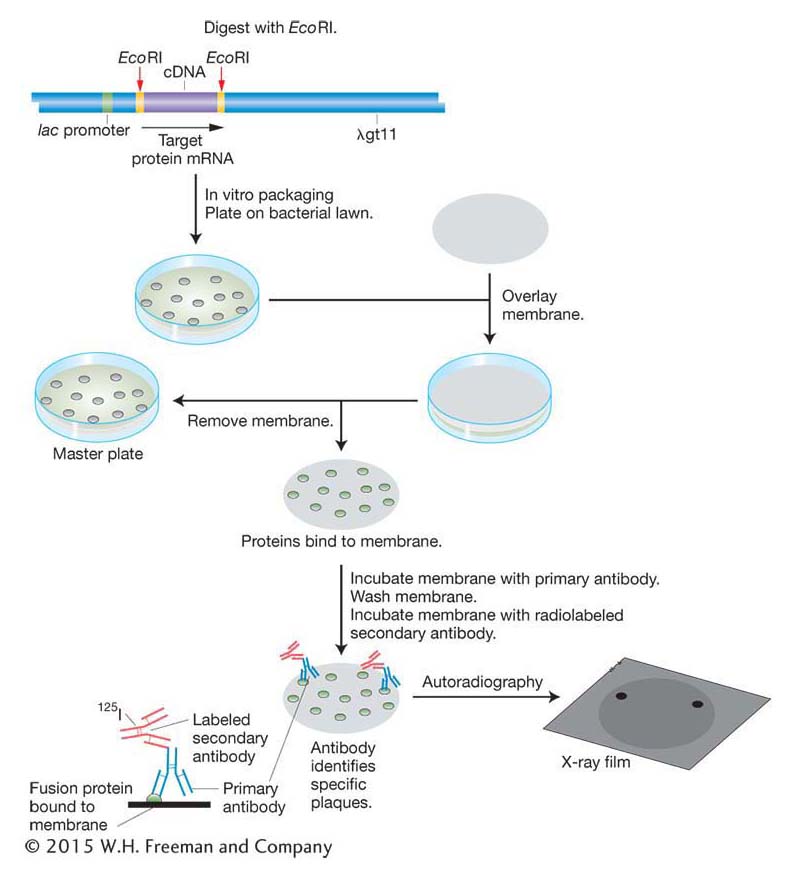
We can see how this type of probe works in practice by returning to the human insulin example. To clone a cDNA corresponding to human insulin, we first synthesize cDNA using mRNA isolated from pancreas cells as the template. The cDNA molecules are then inserted into a bacterial expression vector and the vector is transformed into bacteria. Bacterial colonies containing insulin cDNA will express insulin protein. The insulin protein is identified by its binding with an insulin antibody as described above.
Finding specific clones by functional complementation
In many cases, we don’t have a probe for the gene to start with, but we do have a recessive mutation in the gene of interest. This gene could be a mutant gene in a bacterium or yeast or even a plant or mouse. The goal of this approach is to identify the clone containing the gene of interest by the fact that it will restore the function eliminated by the recessive mutation. In practice, one first generates a genomic or cDNA library from an organism that has the wild-
370
Make a library containing wild-
type a+ recombinant- donor DNA inserts. Transform cells of recessive-
mutant- cell- line a− with this library of DNA inserts. Identify clones from the library that produce transformed cells with the dominant a+ phenotype.
Recover the a+ gene from the successful bacterial or phage clone.
So far, we have described techniques to transform only bacterial cells. You will see later in this chapter that DNA can be introduced into many genetic model organisms, including Saccharomyces cerevisiae (yeast), Caenorhabditis elegans (nematode worm), Arabidopsis thaliana (plant), and Mus musculus (mouse).
371
KEY CONCEPT
A cloned gene can be selected from a library by using probes for the gene’s DNA sequence or for the gene’s protein product or by complementing a mutant phenotype.Southern- and Northern-blot analysis of DNA
After you have amplified your PCR product or selected a clone of interest from a genomic or cDNA library, the next step is to find out more about the DNA. Let’s say that you have recovered the insulin cDNA from an expression vector and want to determine the restriction sites in the genomic copy of the insulin gene. Perhaps you want to see whether these sites differ among diverse human populations. You might also want to know whether the size of the insulin mRNA varies among human populations. Alternatively, you might want to determine whether a similar gene is present in the genome of a related organism. In the section below, you will see that these important questions can be answered by using relatively simple techniques. In these techniques, complex mixtures of DNA or RNA are sorted by size and then probed by hybridization to detect DNA molecules related to some other DNA molecule.
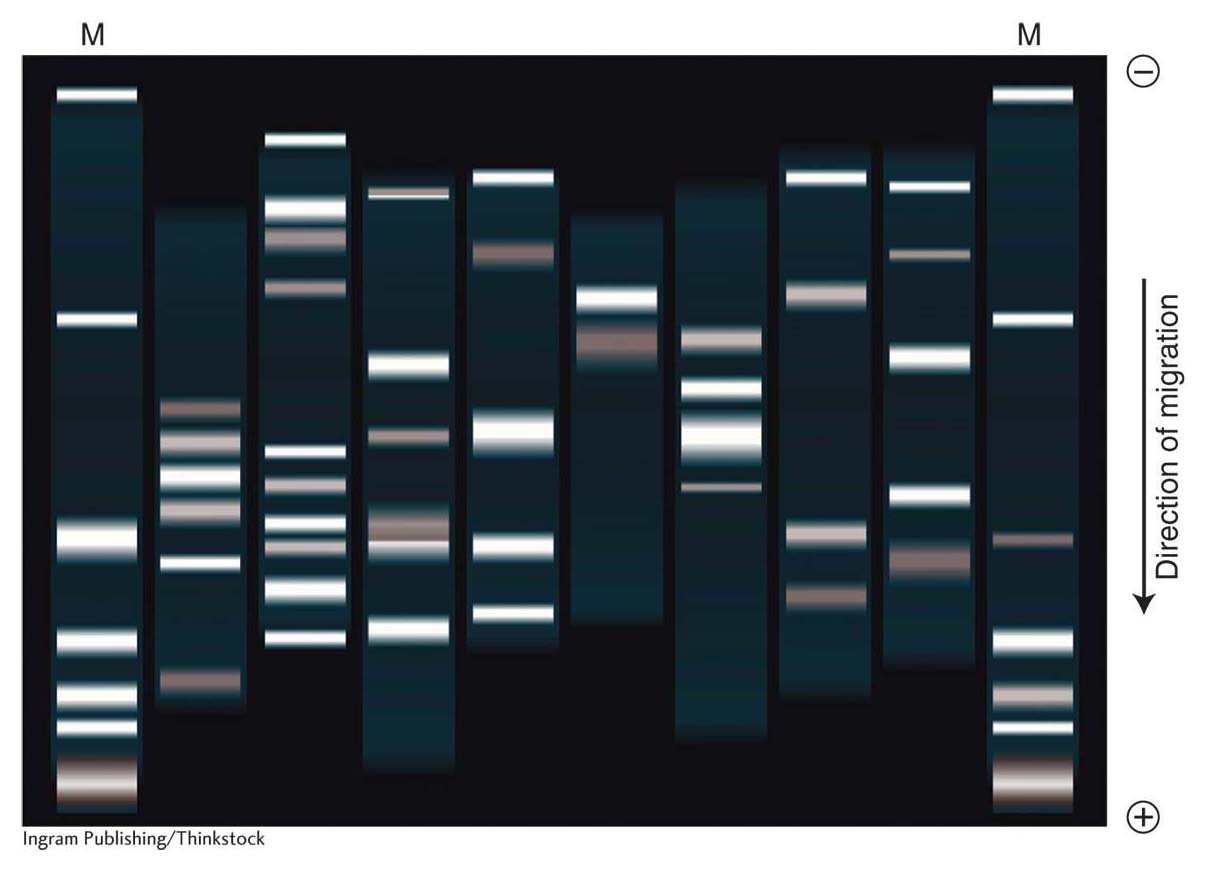
The most extensively used method for detecting a molecule within a mixture is blotting, which starts with gel electrophoresis to separate the molecules in the mixture. A mixture of linear DNA molecules is placed into a well formed in an agarose gel. The gel is oriented in a box with electrodes at either end so that the wells are at the cathode end (negatively charged) and the DNA, because of its negative charge, migrates to the anode end (positively charged). The speed of migration of DNA molecules in the gel is inversely dependent on their size because the agarose acts as a sieve through which small molecules move more easily and quickly than larger fragments (Figure 10-14). Therefore, the fragments in distinct size classes will form distinct bands on the gel. The bands can be visualized by staining the DNA with ethidium bromide, which causes the DNA to fluoresce in ultraviolet light. The absolute size of each fragment in the mixture can be determined by comparing its migration distance with a set of standard fragments of known sizes. If the bands are well separated, an individual band can be cut from the gel, and the DNA sample can be purified from the gel matrix. Therefore, DNA electrophoresis can be either diagnostic (showing sizes and relative amounts of the DNA fragments present) or preparative (useful in isolating specific DNA fragments).
372
Genomic DNA digested by restriction enzymes generally yields so many fragments that electrophoresis produces a continuous smear of DNA and no discrete bands. A probe can identify one fragment in this mixture, with the use of a technique developed by E. M. Southern called Southern blotting (Figure 10-15a). Like clone identification (see Figure 10-12), this technique entails getting an imprint of DNA molecules on a membrane by using the membrane to blot the gel after electrophoresis is complete. The DNA must be denatured first, which allows it to stick to the membrane. Then the membrane is hybridized with a labeled probe. An autoradiogram or a photograph of fluorescent bands will reveal the presence of any bands on the gel that are complementary to the probe. To detect the insulin gene, we can apply this protocol to human genomic DNA digested with restriction enzymes on the membrane, using insulin cDNA as the labeled probe.
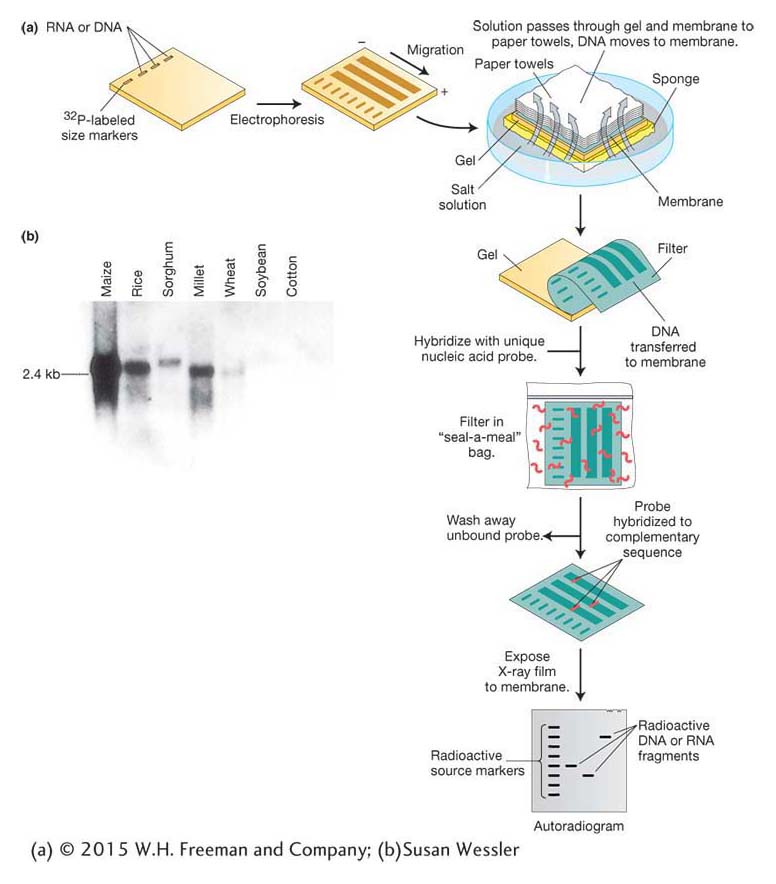
The Southern-
We started this section on blot analysis by posing questions about the insulin gene and its mRNA in human populations and in related species. Based on the techniques above, can you design Southern-
Hence, we see that cloned DNA finds widespread application as a probe used for detecting a specific clone, a DNA fragment, or an RNA molecule. In all these cases, note that the technique again exploits the ability of nucleic acids with complementary nucleotide sequences to find and bind to each other.
KEY CONCEPT
Recombinant-Probing for a specific protein Probing for proteins is generally performed by using antibodies as probes. An antibody is a protein made by an animal’s immune system; it binds with high affinity to a molecule such as a specific protein (which acts as an antigen) because the antibody has a specific lock-
373
374