12.3 Dynamic Chromatin
A second mechanism for influencing gene transcription in eukaryotes modifies the local chromatin structure around gene regulatory sequences. To fully understand how this mechanism works, we need to first understand chromatin structure and then consider how it can change and how these changes affect gene expression.
443
The recruitment of transcriptional machinery by activators may appear to be somewhat similar in eukaryotes and bacteria, with the major difference being the number of interacting proteins in the transcriptional machinery. Indeed, two decades ago, many biologists pictured eukaryotic regulation simply as a biochemically more complicated version of what had been discovered in bacteria. However, this view has changed dramatically as biologists have considered the effect of the organization of genomic DNA in eukaryotes.
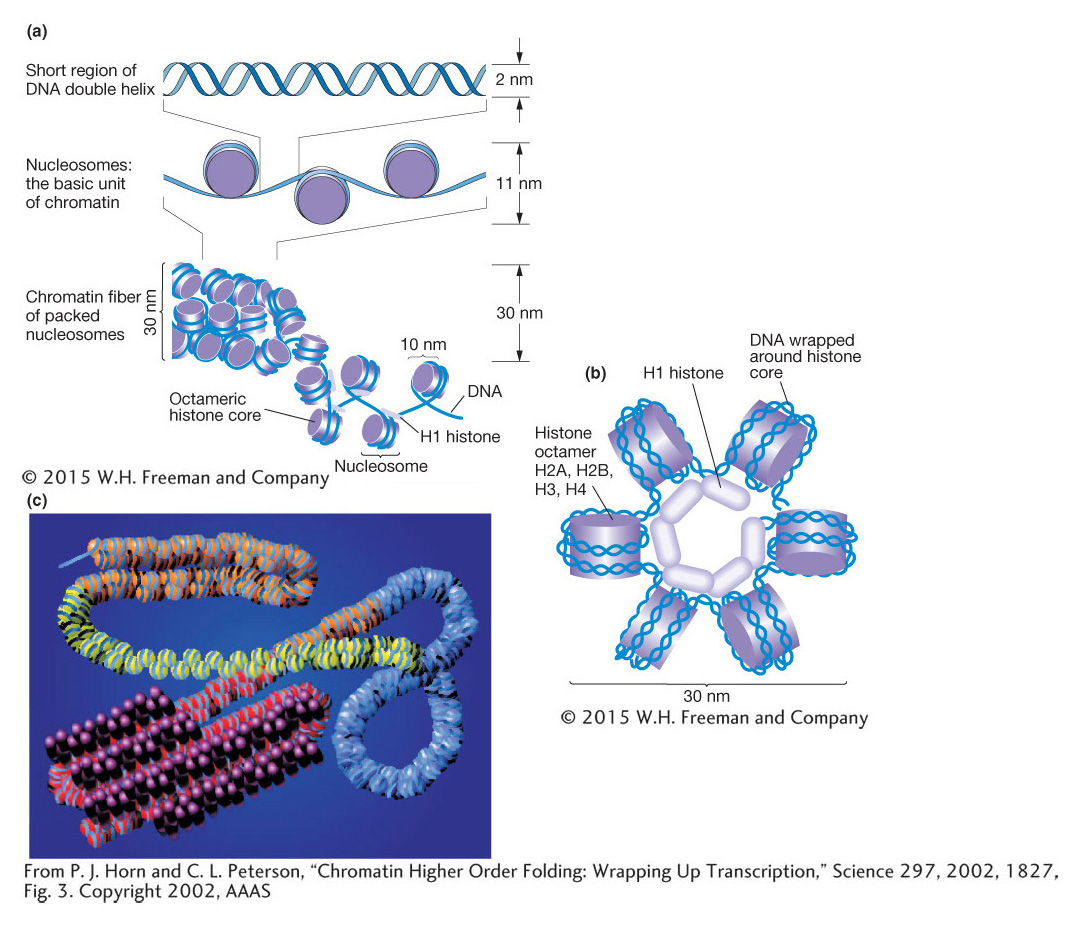
Compared with eukaryotic DNA, bacterial DNA is relatively “naked,” making it readily accessible to RNA polymerase. In contrast, eukaryotic chromosomes are packaged into chromatin, which is composed of DNA and proteins (mostly histones). The basic unit of chromatin is the nucleosome, which contains ~150 bp of DNA wrapped 1.7 times around a core of histone proteins (Figure 12-11). The nucleosome core contains eight histones, two subunits of each of the four histones: histones 2A, 2B, 3, and 4 (called H2A, H2B, H3, and H4) organized as two dimers of H2A and H2B and a tetramer of H3 and H4. Surrounding the nucleosome core is a linker histone, H1, which can compact the nucleosomes into higher-
444
The packaging of eukaryotic DNA into chromatin means that much of the DNA is not readily accessible to regulatory proteins and the transcriptional apparatus. Thus, whereas prokaryotic genes are generally accessible and “on” unless repressed, eukaryotic genes are inaccessible and “off” unless activated. Therefore, the modification of chromatin structure is a distinctive feature of many eukaryotic processes including gene regulation (discussed in this chapter), DNA replication (Chapter 7), and DNA repair (Chapter 16). There are three major mechanisms to alter chromatin structure that will be discussed in depth in this section:
moving nucleosomes along the DNA, also called chromatin remodeling.
histone modification in the nucleosome core.
replacing the common histones in a nucleosome with histone variants.
Chromatin-remodeling proteins and gene activation
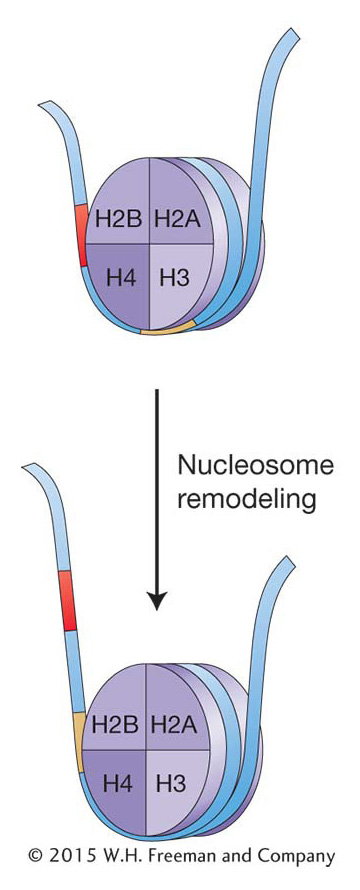
One way to alter chromatin structure might be simply to move the histone octamer along the DNA. In the 1980s, biochemical techniques were developed that allowed researchers to determine the position of nucleosomes in and around specific genes. In these studies, chromatin was isolated from tissues or cells in which a gene was on and compared with chromatin from tissue where the same gene was off. The result for most genes analyzed was that nucleosome positions changed, especially in a gene’s regulatory regions. Thus, which DNA regions are wrapped up in nucleosomes can change: nucleosome positions can shift on the DNA from cell to cell and over the life cycle of an organism. Transcription is repressed when the promoter and flanking sequences are wound up in a nucleosome, which prevents the initiation of transcription by RNA pol II. Activation of transcription would thus require nudging the nucleosomes away from the promoter. Conversely, when gene repression is necessary, nucleosomes shift into a position that prevents transcription. The changing of nucleosome position is referred to as chromatin remodeling. Chromatin remodeling is known to be an integral part of eukaryotic gene expression, and great advances are being made in determining the underlying mechanism(s) and the regulatory proteins taking part. Here, again, genetic studies in yeast have been pivotal.
Two genetic screens in yeast for mutants in seemingly unrelated processes led to the discovery of the same gene whose product plays a key role in chromatin remodeling. In both cases, yeast cells were treated with agents that would cause mutations. In one screen, these mutagenized yeast cells were screened for cells that could not grow well on sucrose (sugar nonfermenting mutants, snf). In another screen, mutagenized yeast cells were screened for mutants that were defective in switching their mating type (switch mutants, swi; see Section 12.5). Many mutants for different loci were recovered in each screen, but one mutant gene was found to cause both phenotypes. Mutants at the so-
What was the connection between the ability to utilize sugar and the ability to switch mating types? The Swi2-
Gal4 also binds to the SWI–
445
A second reason is that many transcription factors act in combinations to control gene expression synergistically. We will see shortly that this combinatorial synergy is a result of the fact that chromatin-
KEY CONCEPT
Chromatin can be dynamic; nucleosomes are not necessarily in fixed positions on the chromosome. Chromatin remodeling changes nucleosome density or position and is an integral part of eukaryotic gene regulation.Modification of histones
Let’s look at the nucleosome more closely to see if any part of this structure could carry the information necessary to influence nucleosome position, nucleosome density, or both.
As already stated, most nucleosomes are composed of a histone octamer made up of two dimers of H2A and H2B and a tetramer of H3 and H4. Histones are known to be the most conserved proteins in nature; that is, histones are almost identical in all eukaryotic organisms from yeast to plants to animals. In the past, this conservation contributed to the view that histones could not take part in anything more complicated than the packaging of DNA to fit in the nucleus. However, recall that DNA with just its four bases was once considered too simple a molecule to carry the blueprint for all organisms on Earth.
Figure 12-13a shows a model of nucleosome structure that represents contributions from many studies. Of note is that the histone proteins are organized into the core octamer with some of their amino-
446
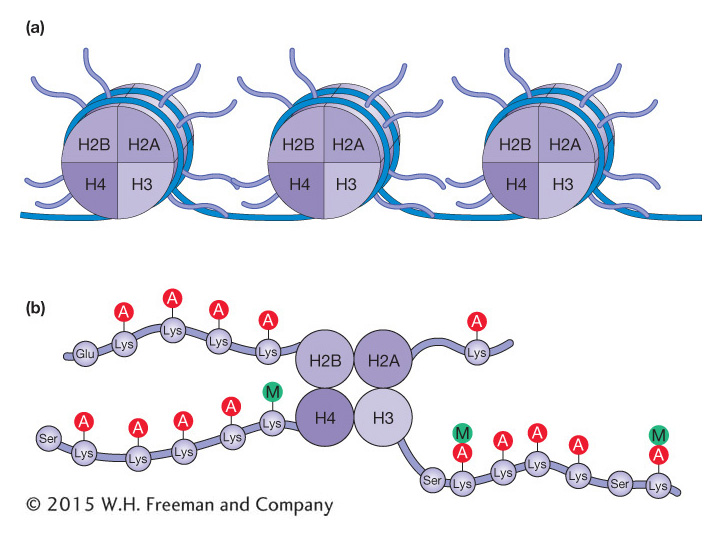
There are now known to be at least 150 different histone modifications that utilize a wide variety of molecules in addition to the acetyl and methyl groups already mentioned, including phosphorylation, ubiquitination, and ADP ribosylation. The covalent modification of histone tails is said to contribute to a histone code. Scientists coined the expression “histone code” because the covalent modification of histone tails is reminiscent of the genetic code. For the histone code, information is stored in the patterns of histone modification rather than in the sequence of nucleotides. With more than 150 known histone modifications, there are a huge number of possible patterns, and scientists are just beginning to decipher their effects on chromatin structure and transcriptional regulation. To add to this complexity, the code is likely not interpreted in precisely the same way in all organisms. The role of histone acetylation and methylation in gene expression is described below.
Histone acetylation, deacetylation, and gene expression The acetylation reaction is one of the best-
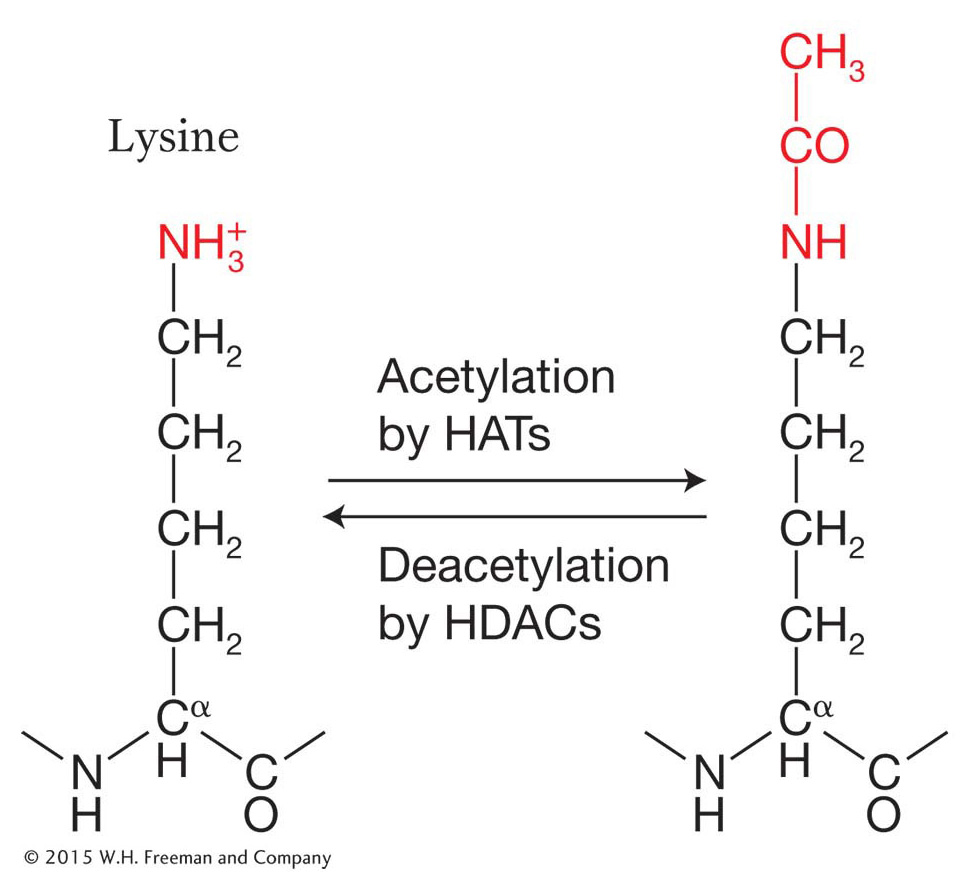
Note that the reaction is reversible, which means that acetyl groups can be added by the enzyme histone acetyltransferase (HAT) and removed by the enzyme histone deacetylase (HDAC) from the same histone residue. For now, let’s see how the acetylation and deacetylation of histone amino acids influences chromatin structure and gene expression.
Evidence had been accumulating for years that the histones associated with the nucleosomes of active genes are rich in acetyl groups (said to be hyperacetylated), whereas inactive genes are underacetylated (hypoacetylated). The HAT enzyme proved very difficult to isolate. When it was finally isolated and its protein sequence deduced, it was found to be an ortholog of a yeast transcriptional activator called GCN5 (meaning that it was encoded by the same gene in a different organism). Thus, the conclusion was that GCN5 is a histone acetyltransferase. It binds to the DNA in the regulatory regions of some genes and activates transcription by acetylating nearby histones. Various protein complexes that are recruited by transcriptional activators are now understood to possess HAT activity.
How does histone acetylation alter chromatin structure and, in the process, facilitate changes in gene expression? The addition of acetyl groups to histone residues neutralizes the positive charge of lysine residues and reduces the interaction of the histone tails with the negatively charged DNA backbone. This results in more open chromatin as the electrostatic interactions between adjacent nucleosomes and between nucleosomes and adjacent DNA are reduced (Figure 12-14). In addition, histone acetylation, in conjunction with other histone modifications, influences the binding of regulatory proteins to the DNA. A bound regulatory protein may take part in one of several functions that either directly or indirectly increase the frequency of transcription initiation.
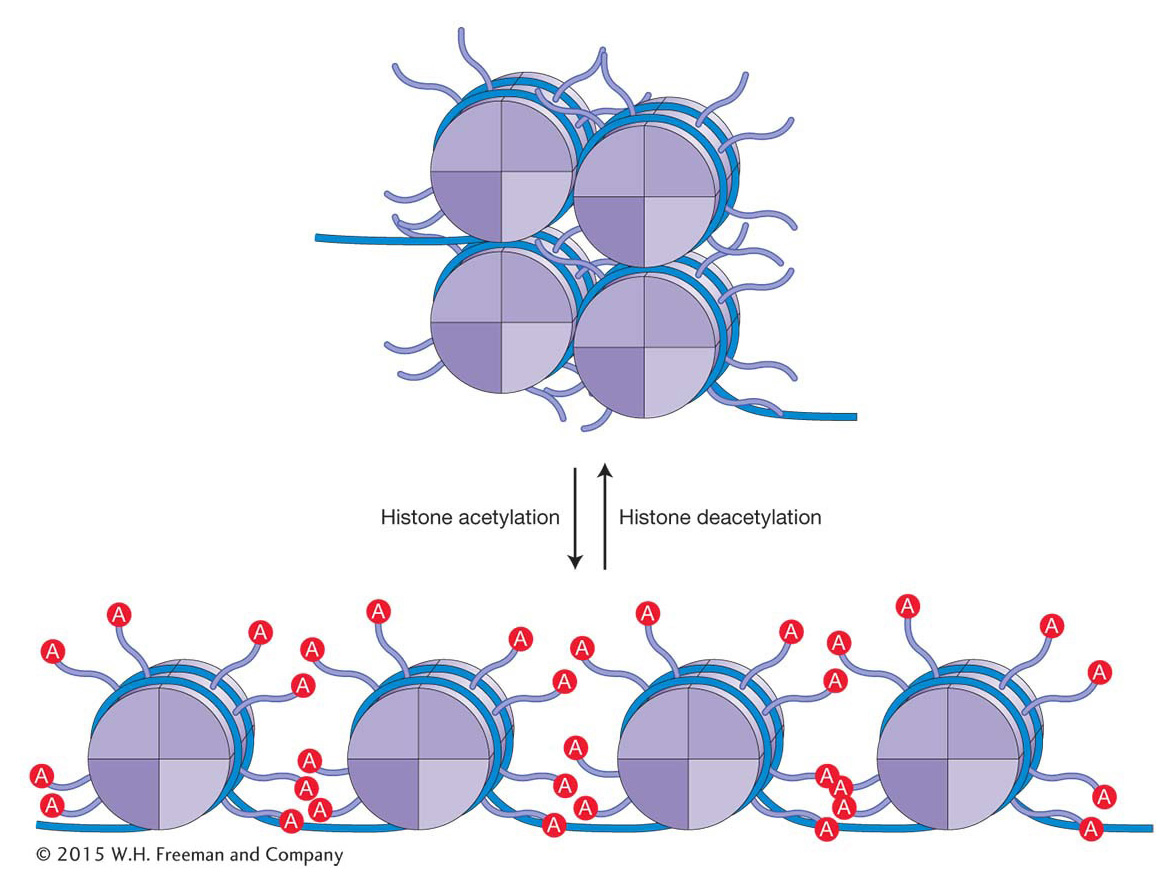
447
Like other histone modifications, acetylation is reversible, and HDACs play key roles in gene repression. For example, in the presence of galactose and glucose, the activation of GAL genes is prevented by the Mig1 protein. Mig1 is a sequence-
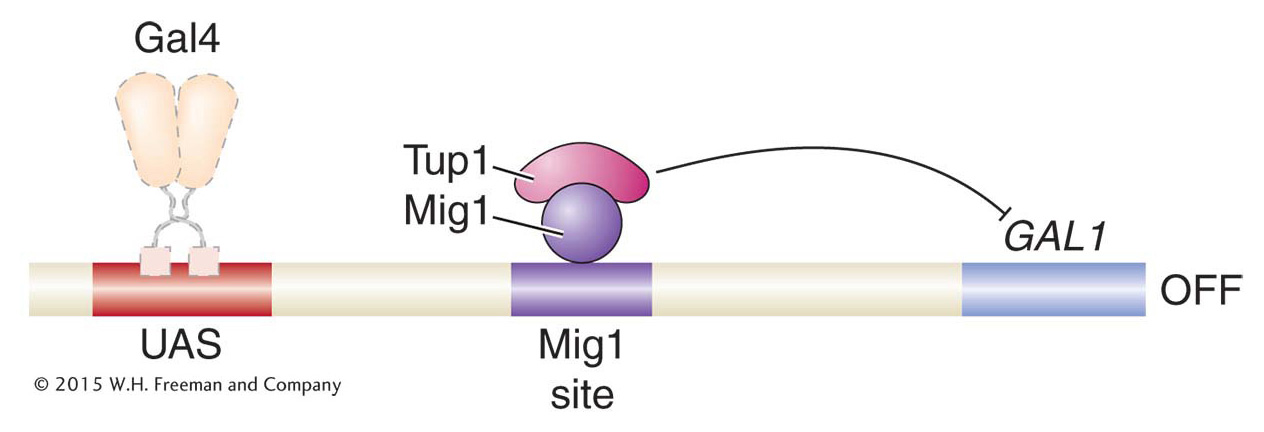
448
Histone methylation can activate or repress gene expression
Methylation is another post-
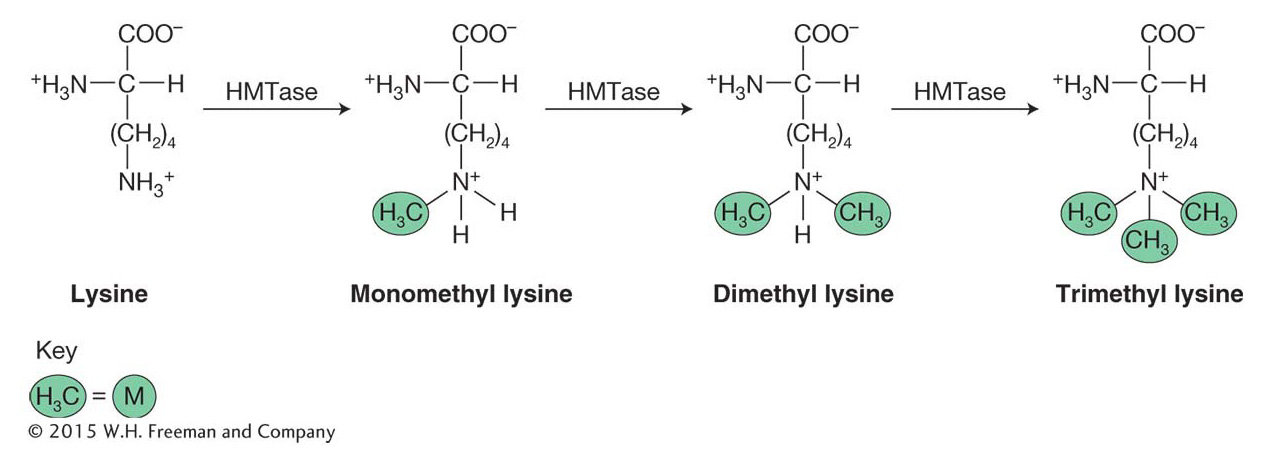
The amino acid lysine is abbreviated with a “K.” As such, these post-
Unlike acetylation, the addition of methyl groups can either activate or repress gene expression. Recall that acetylation of lysine acts to neutralize the positive histone charge and, in this direct way, activates gene expression by reducing interactions between nucleosomes and DNA, opening chromatin. In contrast, methylation of specific lysine residues, which does not affect charge, creates binding sites for other proteins that either activate or repress gene expression depending on the residues modified. For example, methylation of H3 lysine residue 4 [H3K4(me)] is associated with the activation of gene expression and is enriched in nucleosomes near the start of transcription. There is a very different outcome when H3K9 or H3K27 are methylated. These modifications, which are associated with gene repression and tightly packed chromatin, will be discussed in greater detail later in this chapter.
KEY CONCEPT
Post-The inheritance of histone modifications and chromatin structure
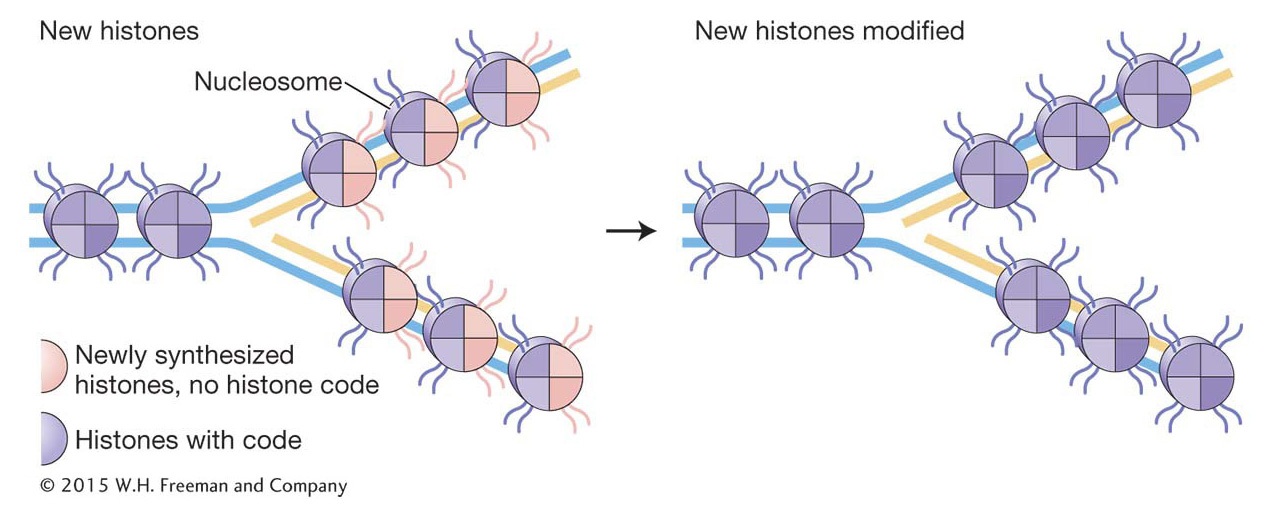
An important feature of chromatin structure is that it can be inherited. This form of inheritance is given a name—
Recall that prokaryotic replication is orchestrated at the replication fork by the replisome, a molecular machine that includes two DNA pol III holoenzymes and accessory proteins (see Figure 7-
449
KEY CONCEPT
The eukaryotic replisome performs all the functions of the prokaryotic replisome; in addition, it must disassemble and reassemble the protein–Histone variants
Unlike the common (also called consensus) histones that are added during DNA replication, eukaryotes also have other histones, called histone variants, that can replace the consensus histones that have already been assembled into nucleosomes. For example, two variants for histone H2 are called H2A.Z and H2A.X, and one H3 variant is called CENP-
DNA methylation: another heritable mark that influences chromatin structure
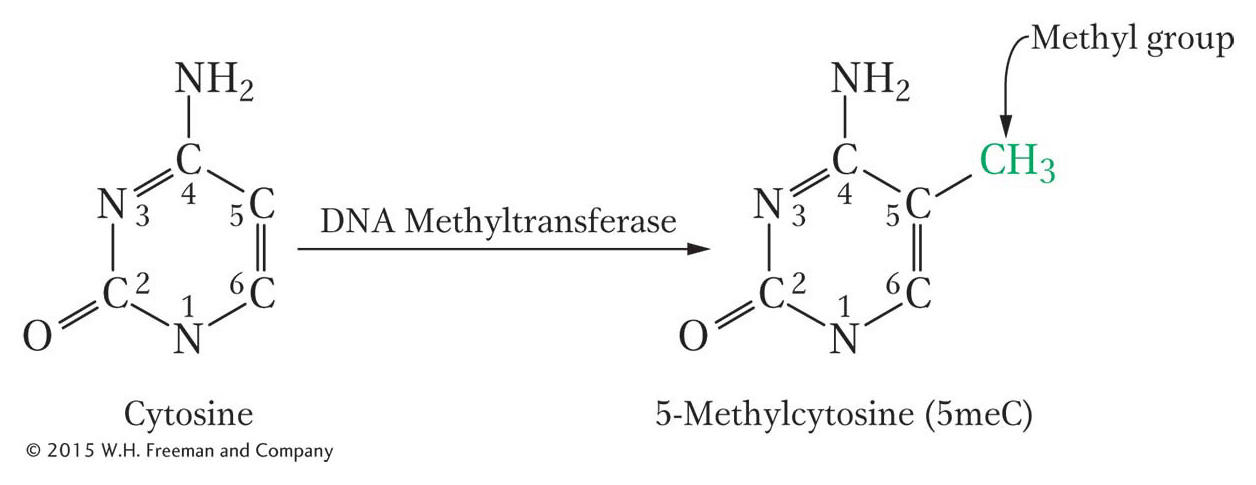
There is another important epigenetic mark in most (but not all) eukaryotes. This mark is not a histone modification; rather, it is the addition of methyl groups to DNA residues after replication. An enzyme usually attaches these methyl groups to the carbon-
450
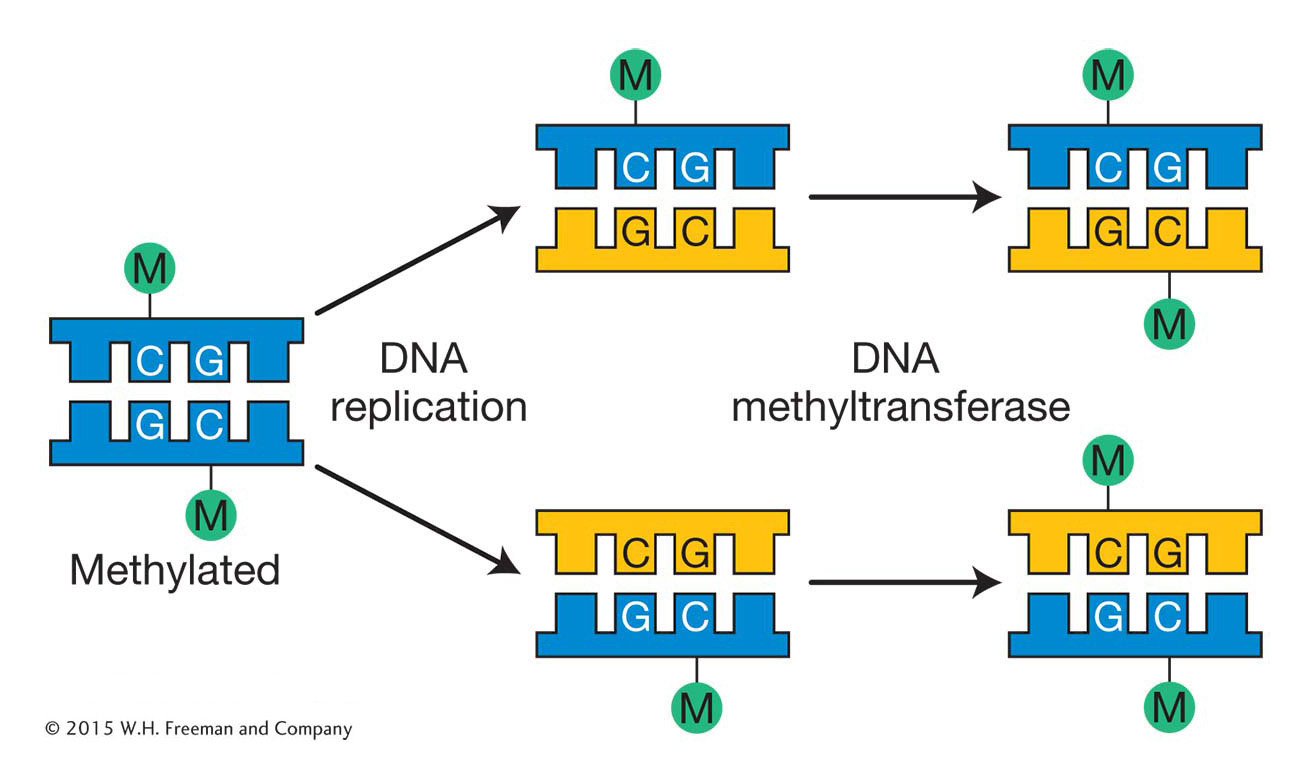
In mammals, the methyl group is usually added to the cytosine in a CG dinucleotide. The pattern of methylation is called symmetric methylation because the methyl groups are present on both strands in the same context:
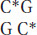
A remarkable number of C residues are methylated in mammals: 70 to 80 percent of all CG dinucleotides are methylated genome-
Like histone modifications, DNA methylation marks can be stably inherited from one cell generation to the next. The inheritance of DNA methylation is better understood than the inheritance of histone modifications. Semiconservative replication generates daughter helices that are methylated on only the parental strand. DNA molecules methylated on only one strand are termed hemimethylated. Methyl groups are added to unmethylated strands by DNA methyltransferases that have a high affinity for these hemimethylated substrates. These enzymes are guided by the methylation pattern on the parental strand (Figure 12-17). As you will see later in the chapter, because DNA methylation is more stable than histone modifications, it is often associated with regions of the genome that are maintained in an inactive state for the entire lifetime of an organism. Such regions will be discussed later in this chapter.