14.2 Obtaining the Sequence of a Genome
When people encounter new territory, one of their first activities is to create a map. This practice has been true for explorers, geographers, oceanographers, and astronomers, and it is equally true for geneticists. Geneticists use many kinds of maps to explore the terrain of a genome. Examples are linkage maps based on inheritance patterns of gene alleles and cytogenetic maps based on the location of microscopically visible features such as rearrangement break points.
The highest-
Turning sequence reads into an assembled sequence
You’ve probably seen a magic act in which the magician cuts up a newspaper page into a great many pieces, mixes it in his hat, says a few magic words, and voila! an intact newspaper page reappears. Basically, that’s how genomic sequences are obtained. The approach is to (1) break the DNA molecules of a genome up into thousands to millions of more or less random, overlapping small segments; (2) read the sequence of each small segment; (3) computationally find the overlap among the small segments where their sequences are identical; and (4) continue overlapping ever larger pieces until all the small segments are linked (Figure 14-2). At that point, the sequence of a genome is assembled.
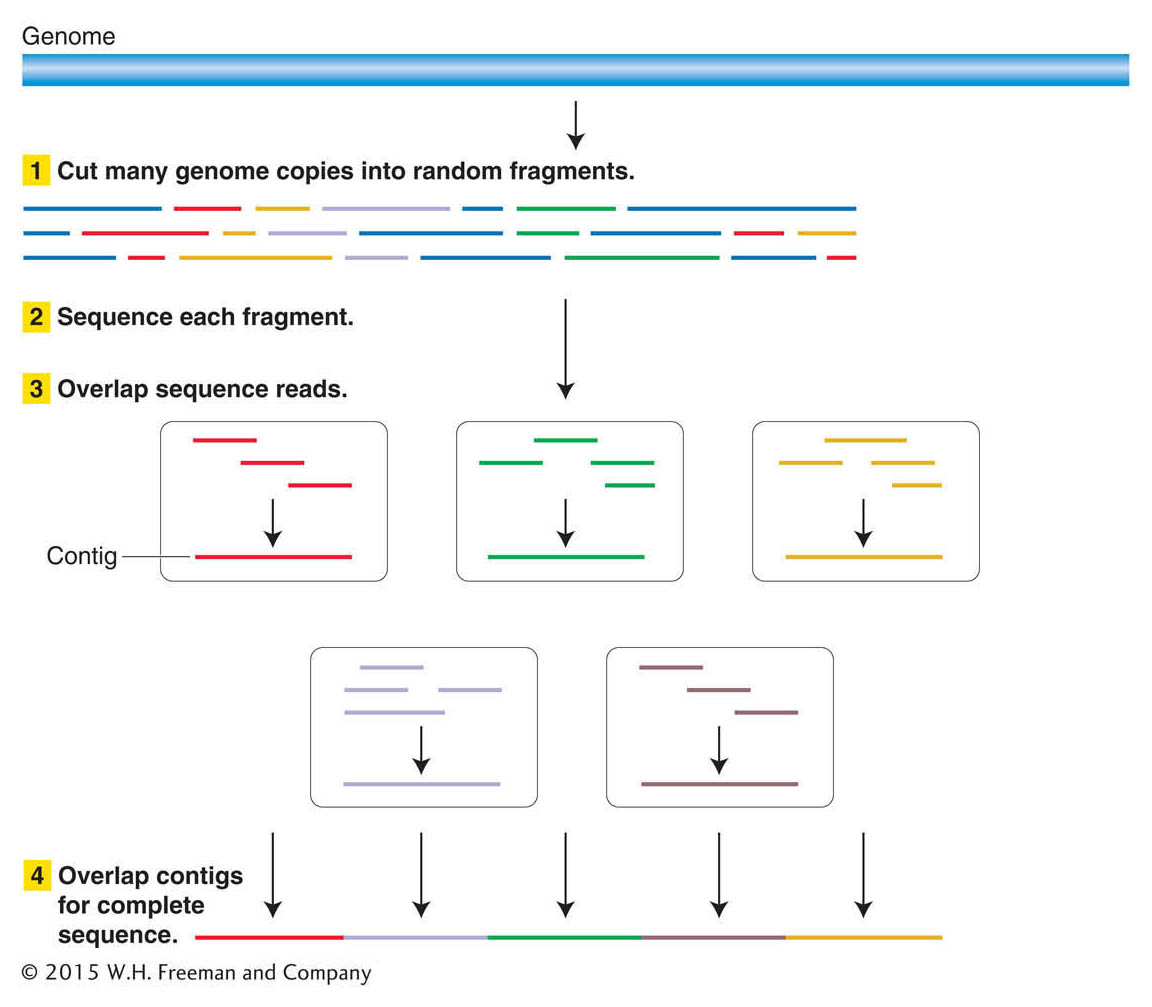
Why does this process require automation? To understand why, let’s consider the human genome, which contains about 3 × 109 bp of DNA, or 3 billion base pairs (3 gigabase pairs = 3 Gbp). Suppose we could purify the DNA intact from each of the 24 human chromosomes (X, Y, and the 22 autosomes), separately put each of these 24 DNA samples into a sequencing machine, and read their sequences directly from one telomere to the other. Obtaining a complete sequence would be utterly straightforward, like reading a book with 24 chapters—
512
Rather, automated sequencing is the current state of the art in DNA sequencing technology. Initially based on the pioneering Sanger dideoxy chain-
Individual sequencing reactions (called sequencing reads) provide letter strings that, depending on the sequencing technique employed, range on average from about 100 to 5000 bases long. Such lengths are tiny compared with the DNA of a single chromosome. For example, an individual read of 300 bases is only 0.0001 percent of the longest human chromosome (about 3 × 108 bp of DNA) and only about 0.00001 percent of the entire human genome. Thus, one major challenge facing a genome project is sequence assembly—that is, building up all of the individual reads into a consensus sequence, a sequence for which there is consensus (or agreement) that it is an authentic representation of the sequence for each of the DNA molecules in that genome.
Let’s look at these numbers in a somewhat different way to understand the scale of the problem. As with any experimental observation, automated sequencing machines do not always give perfectly accurate sequence reads. Indeed, newer, higher-
513
Given an average sequence read of about 100 bases of DNA and a human genome of 3 billion base pairs, 300 million independent reads are required to give 10-
What are the goals of sequencing a genome? First, we strive to produce a consensus sequence that is a true and accurate representation of the genome, starting with one individual organism or standard strain from which the DNA was obtained. This sequence will then serve as a reference sequence for the species. We now know that there are many differences in DNA sequence between different individuals within a species and even between the maternally and paternally contributed genomes within a single diploid individual. Thus, no one genome sequence truly represents the genome of the entire species. Nonetheless, the genome sequence serves as a standard or reference with which other sequences can be compared, and it can be analyzed to determine the information encoded within the DNA, such as the inventory of encoded RNAs and polypeptides.
Like written manuscripts, genome sequences can range from draft quality (the general outline is there, but there are typographical errors, grammatical errors, gaps, sections that need rearranging, and so forth), to finished quality (a very low rate of typographical errors, some missing sections but everything that is currently possible has been done to fill in these sections), to truly complete (no typographical errors, every base pair absolutely correct from telomere to telomere). In the following sections, we will examine the strategy and some methods for producing draft and finished genome-
Whole-genome sequencing
The current general strategy for obtaining and assembling the sequence of a genome is called whole-
Traditional WGS
The traditional WGS approach begins with the construction of genomic libraries, which are collections of these short segments of DNA, representing the entire genome. The short DNA segments in such a library have been inserted into one of a number of types of accessory chromosomes (nonessential elements such as plasmids, modified bacterial viruses, or artificial chromosomes) and propagated in microbes, usually bacteria or yeast. These accessory chromosomes carrying DNA inserts are called vectors.
514
To generate a genomic library, a researcher first uses restriction enzymes, which cleave DNA at specific sequences, to cut up purified genomic DNA. Some enzymes cut the DNA at many places, whereas others cut it at fewer places; so the researcher can control whether the DNA is cut, on average, into longer or shorter pieces. The resulting fragments have short single strands of DNA at both ends. Each fragment is then joined to the DNA molecule of the accessory chromosome, which also has been cut with a restriction enzyme and which has ends that are complementary to those of the genomic fragments. In order for the entire genome to be represented, multiple copies of the genomic DNA are cut into fragments. By this means, thousands to millions of different fragment-
The resulting pool of recombinant DNA molecules is then propagated, typically by introducing the molecules into bacterial cells. Each cell takes up one recombinant molecule. Then each recombinant molecule is replicated in the normal growth and division of its host so that many identical copies of the inserted fragment are produced for use in analyzing the fragment’s DNA sequence. Because each recombinant molecule is amplified from an individual cell, each cell is a distinct clone. (More details about DNA cloning are provided in Chapter 10.) The resulting library of clones is called a shotgun library because sequence reads are obtained from clones randomly selected from the whole-
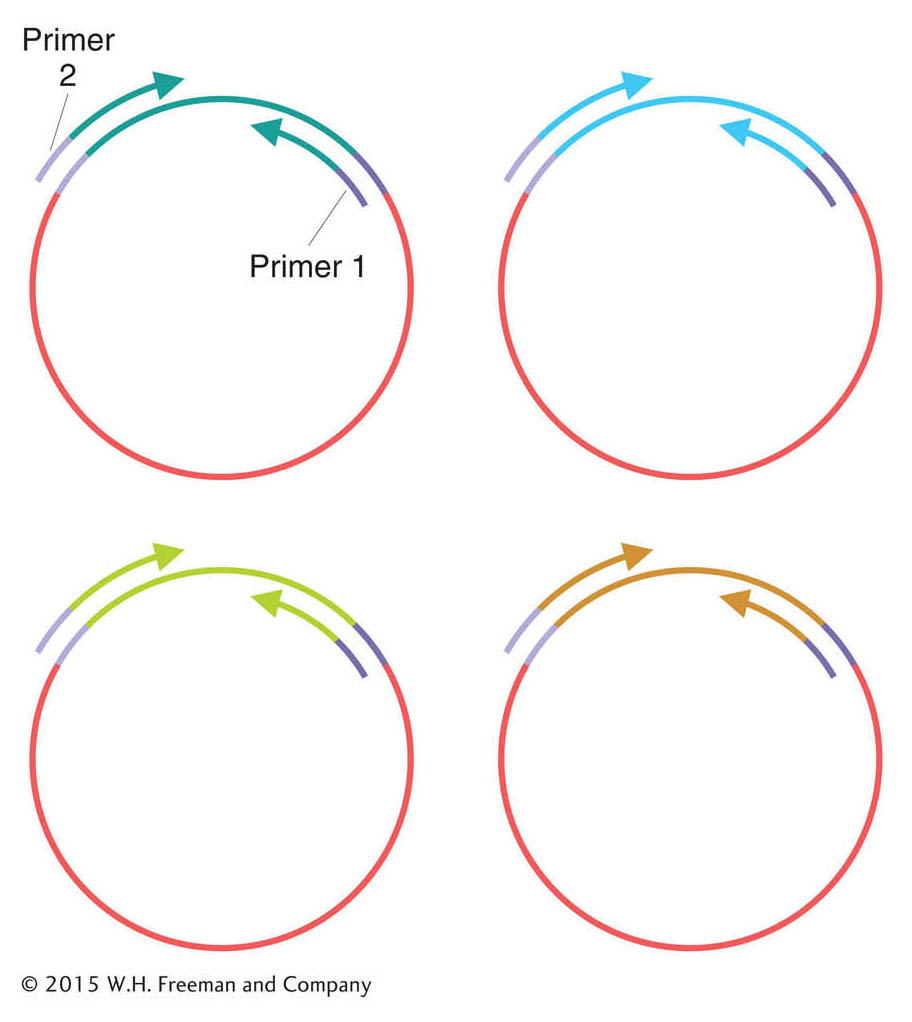
Next, the genome fragments in clones from the shotgun library are partially sequenced. The sequencing reaction must start from a primer of known sequence. Because the sequence of a cloned insert is not known (and is the goal of the exercise), primers are based on the sequence of adjacent vector DNA. These primers are used to guide the sequencing reaction into the insert. Hence, short regions at one or both ends of the genomic inserts can be sequenced (Figure 14-3). After sequencing, the output is a large collection of random short sequences, some of them overlapping. These sequence reads are assembled into a consensus sequence covering the whole genome by matching homologous sequences shared by reads from overlapping clones. The sequences of overlapping reads are assembled into units called sequence contigs (sequences that are contiguous, or touching).
Next-generation whole-genome shotgun sequencing
The goal of next-
- DNA molecules are prepared for sequencing in cell-
free reactions, without cloning in microbial hosts. 515
- Millions of individual DNA fragments are isolated and sequenced in parallel during each machine run.
- Advanced fluid-
handling technologies, cameras, and software make it possible to detect the products of sequencing reactions in extremely small reaction volumes.
Since the field of genomic technology is evolving rapidly, we will not describe every next-
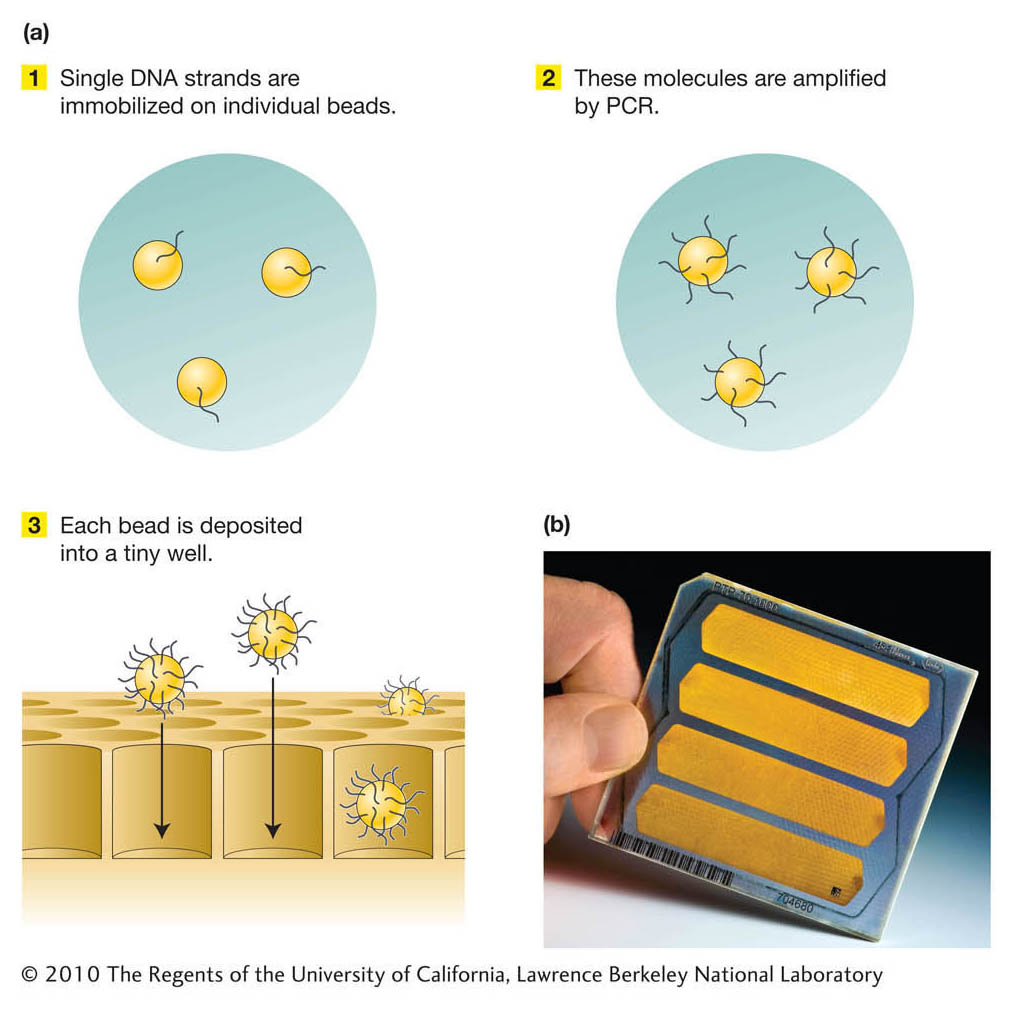
Stage 1. A DNA template library of single-
Stage 2. The DNA molecules in the template library are amplified into many copies, not by growing colonies as for traditional genomic libraries, but by using the polymerase chain reaction (PCR; see Chapter 10). First, single molecules are immobilized on individual beads. The molecules are then amplified by PCR such that single-
516
Stage 3. The sequencing of each bead is performed using a novel “sequencing-
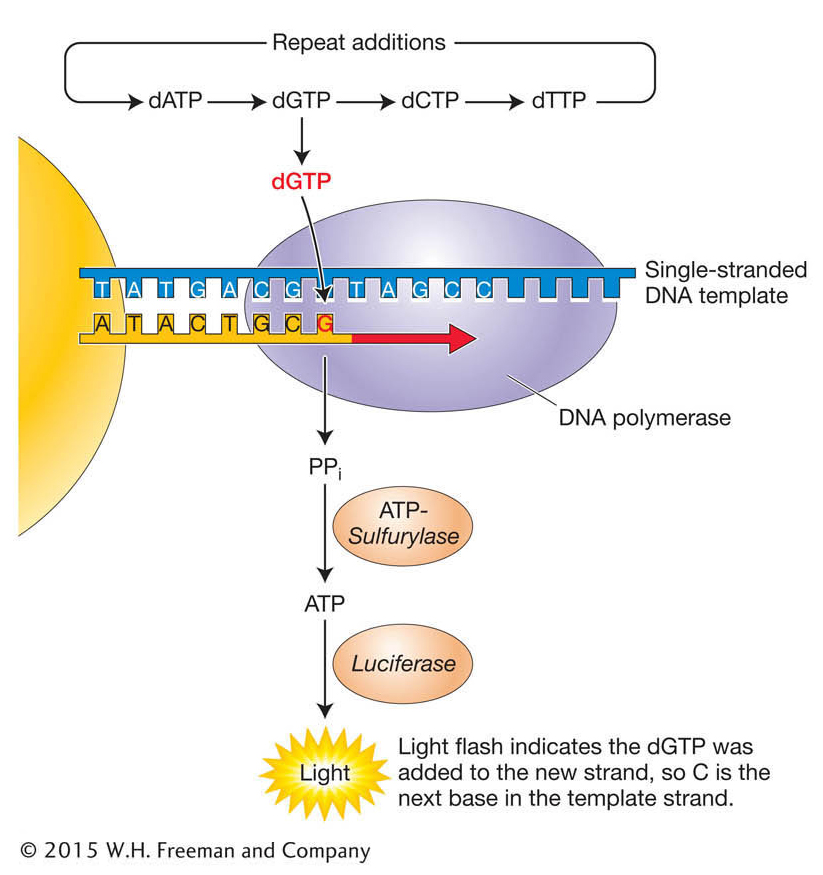
Other widely used platforms such as the Illumina sequencing systems and the Pacific Bioscience systems also detect the synthesis of DNA, but by different means. The Illumina system detects the incorporation of individual, fluorescently labeled dNTPs, while the Pacific Bioscience process detects bases being incorporated into a single, immobilized DNA molecule. The method chosen by investigators depends a great deal on the application. The Illumina system produces a larger number of shorter reads than the 454 system, while the Pacific Bioscience system provides the advantage of much longer individual reads than any other system, but with a higher error rate. The high throughput of each approach is the product of the massively parallel sequencing: several hundred thousand to more than 1 million reactions can be run simultaneously. Earlier sequencing machines were able to achieve just 384 sequencing reactions per run.
517
Whole-genome-sequence assembly
Whichever method of obtaining raw sequence is used, the challenge remains to assemble the contigs into the entire genome sequence. The difficulty of that process depends strongly on the size and complexity of the genome.
For instance, the genomes of bacterial species are relatively easy to assemble. Bacterial DNA is essentially single-
For eukaryotes, genome assembly often presents some difficulties. A big stumbling block is the existence of numerous classes of repeated sequences, some arranged in tandem and others dispersed. Why are they a problem for genome sequencing? In short, because a sequencing read of repetitive DNA fits into many places in the draft of the genome. Not infrequently, a tandem repetitive sequence is in total longer than the length of a maximum sequence read. In that case, there is no way to bridge the gap between adjacent unique sequences. Dispersed repetitive elements can cause reads from different chromosomes or different parts of the same chromosome to be mistakenly aligned together.
KEY CONCEPT
The landscape of eukaryotic chromosomes includes a variety of repetitive DNA segments. These segments are difficult to align as sequence reads.Whole-
The solution to this problem was to make use of the pairs of sequence reads from opposite ends of the genomic inserts in the same clone—
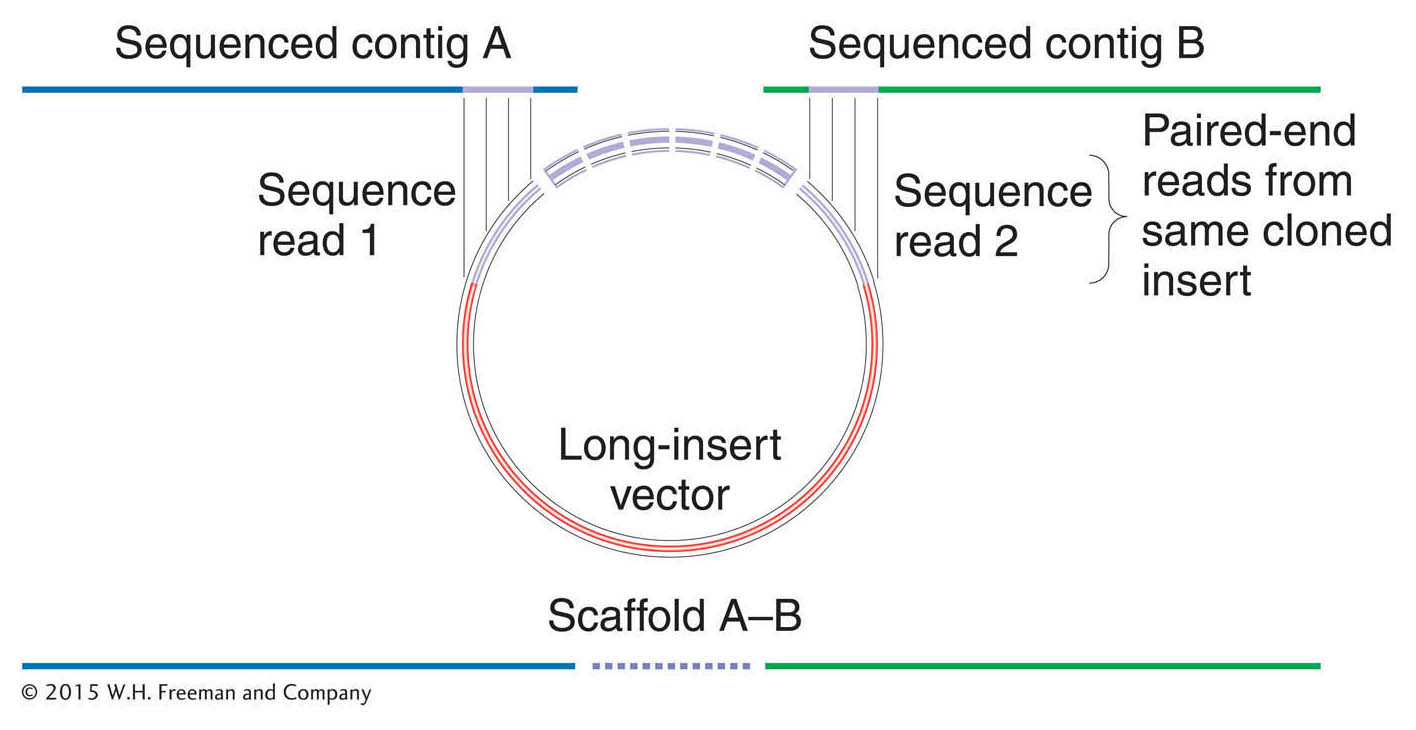
Indeed, because the size of each clone was known (that is, it came from a library containing genomic inserts of uniform size, either the 2-
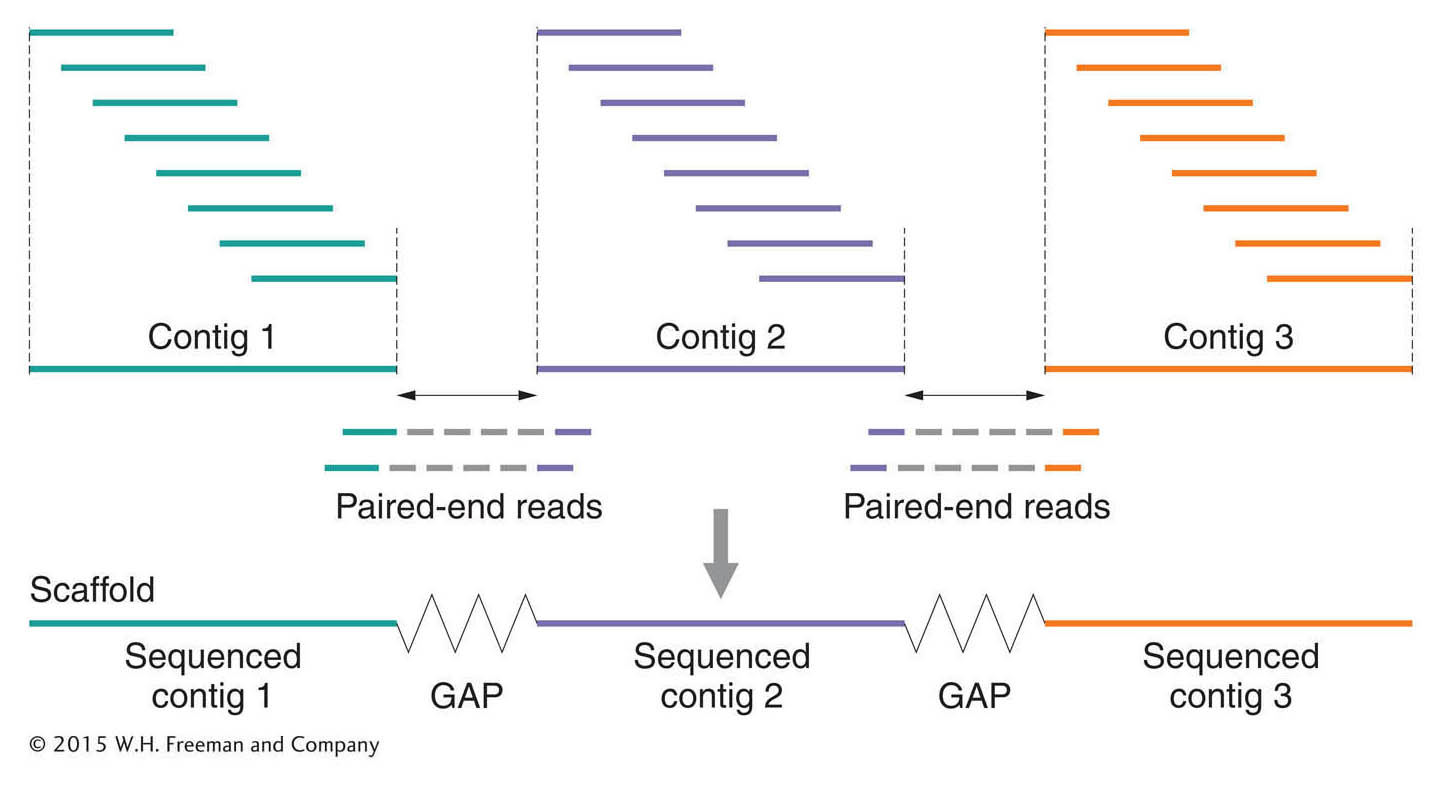
518
Next-
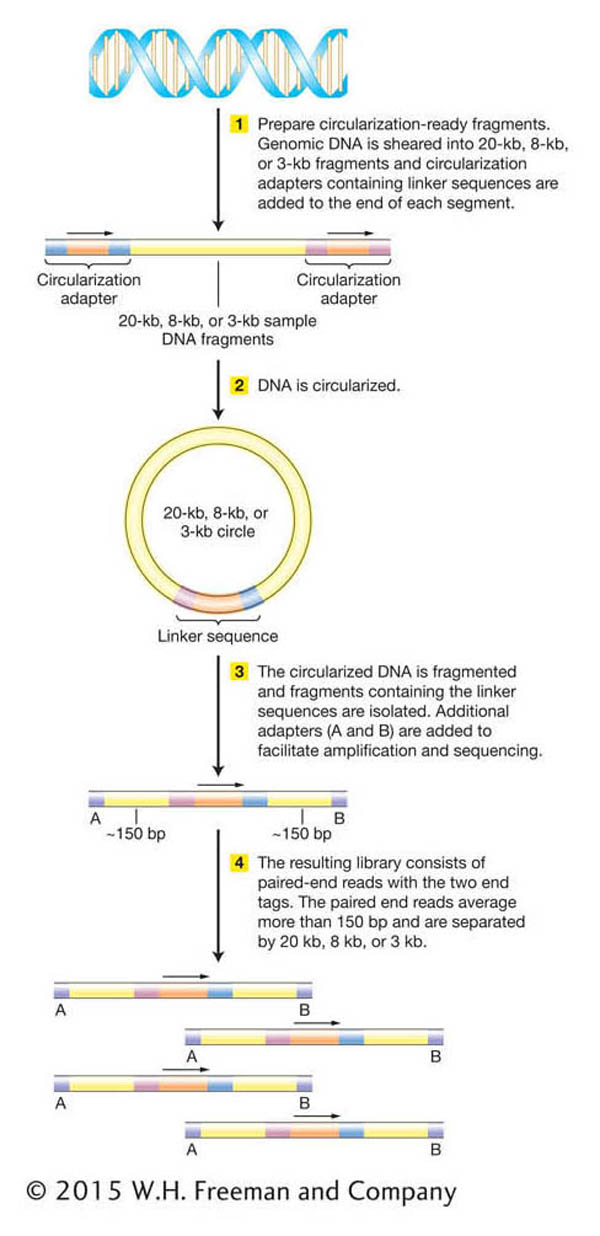
519
In both traditional and next-
Whether a genome is sequenced to “draft” or “finished” standards is a cost–