“’Omics”
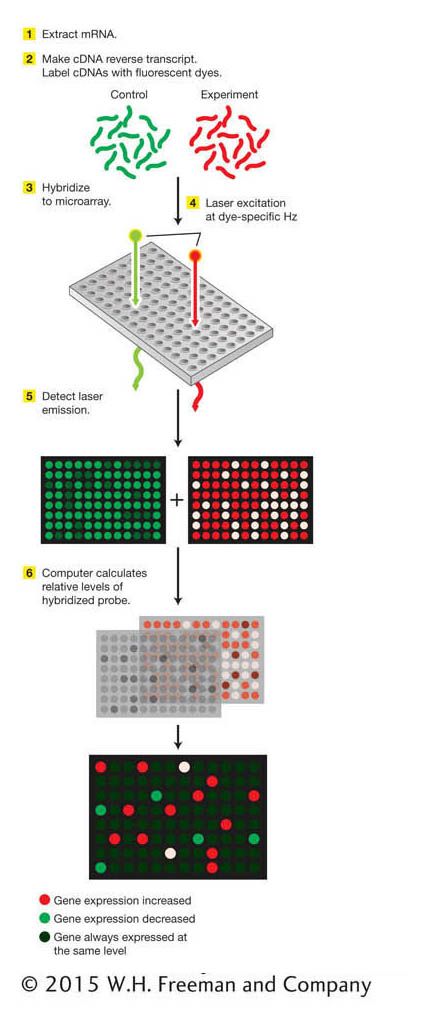
Figure 14-20: Microarrays can detect differences in gene expression
Figure 14-20: The key steps in a microarray analysis are (1) extraction of mRNA from cells or tissues, (2) synthesis of fluorescent-dye-labeled cDNA probes, (3) hybridization to the microarray, (4) detection of the fluorescent signal from hybridized probes, and (5) image analysis to identify relative levels of hybridized probe. The relative levels reveal those genes whose expression is increased or decreased under the conditions analyzed. ANIMATED ART: DNA microarrays: using an oligonucleotide array to analyze patterns of gene expression
In addition to the genome, other global data sets are of interest. Following the example of the term genome, for which “gene” plus “-ome” becomes a word for “all genes,” genomics researchers have coined a number of terms to describe other global data sets on which they are working. This ’ome wish list includes
The transcriptome. The sequence and expression patterns of all RNA transcripts (which kinds, where in tissues, when, how much).
The proteome. The sequence and expression patterns of all proteins (where, when, how much).
The interactome. The complete set of physical interactions between proteins and DNA segments, between proteins and RNA segments, and between proteins.
We will not consider all of these omes in this section but will focus on some of the global techniques that are beginning to be exploited to obtain these data sets.
Using DNA microarrays to study the transcriptome Suppose we want to answer the question, What genes are active in a particular cell under certain conditions? Those conditions could be one or more stages in development, or they could be the presence or absence of a pathogen or a hormone. Active genes are transcribed into RNA, and so the set of RNA transcripts present in the cell can tell us what genes are active. Here is where the new technology of DNA chips used to assay RNA transcripts is so powerful.
DNA chips are samples of DNA laid out as a series of microscopic spots bound to a glass “chip” the size of a microscope cover slip. The set of DNAs so displayed is called a microarray. A typical type of microarray contains short synthetic oligonucleotides representing most or all of the genes in a genome (Figure 14-20). DNA microarrays have powered molecular genetics by permitting the assay of RNA transcripts for all genes simultaneously in a single experiment. Let’s see how this process works in more detail.
Microarrays are exposed to cDNA probes—for example, one set of probes used as a control and one set of probes representing a specific condition. The set used as a control might be made from the total set of RNA molecules extracted from a particular cell type grown under typical conditions. The second set of probes might be made from RNA extracted from cells grown under some experimental condition. Fluorescent labels are attached to the probes, and the probes are hybridized to the microarray. The relative binding of the probe molecules to the microarray is monitored automatically with the use of a laser-beam-illuminated microscope. In this manner, genes whose levels of expression are increased or decreased under the given experimental condition are identified. Similarly, genes that are active in a given cell type or at a given stage of development can be identified.
With an understanding of which genes are active or inactive at a given developmental stage, in a particular cell type, or in various environmental conditions, the sets of genes that may respond to similar regulatory inputs can be identified. Furthermore, gene-expression profiles can paint a picture of the differences between normal and diseased cells. By identifying genes whose expression is altered by mutations, in cancer cells, or by a pathogen, researchers may be able to devise new therapeutic strategies.
Using the two-hybrid test to study the protein–protein interactome One of the most important activities of proteins is their interaction with other proteins. Because of the large number of proteins in any cell, biologists have sought ways of systematically studying all of the interactions of individual proteins in a cell. One of the most common ways of studying the interactome uses an engineered system in yeast cells called the two-hybrid test, which detects physical interactions between two proteins. The basis for the test is the transcriptional activator encoded by the yeast GAL4 gene (see Chapter 12).
Recall that this protein has two domains: (1) a DNA-binding domain that binds to the transcriptional start site and (2) an activation domain that will activate transcription but cannot itself bind to DNA. Thus, the two domains must be in close proximity in order for transcriptional activation to take place. Suppose that you are investigating whether two proteins interact. The strategy of the two-hybrid system is to separate the two domains of the activator encoded by GAL4, making activation of a reporter gene impossible. Each domain is connected to a different protein. If the two proteins interact, they will join the two domains together. The activator will become active and start transcription of the reporter gene.
How is this scheme implemented in practice? The GAL4 gene is divided between two plasmids so that one plasmid contains the part encoding the DNA-binding domain and the other plasmid contains the part encoding the activation domain. On one plasmid, a gene for one protein under investigation is spliced next to the DNA-binding domain, and this fusion protein acts as “bait.” On the other plasmid, a gene for another protein under investigation is spliced next to the activation domain and this fusion protein is said to be the “target” (Figure 14-21). The two hybrid plasmids are then introduced into the same yeast cell—perhaps by mating haploid cells containing bait and target plasmids. The final step is to look for activation of transcription by a GAL4-regulated reporter gene construct, which would be proof that bait and target bind to each other. The two-hybrid system can be automated to make it possible to hunt for protein interactions throughout the proteome.
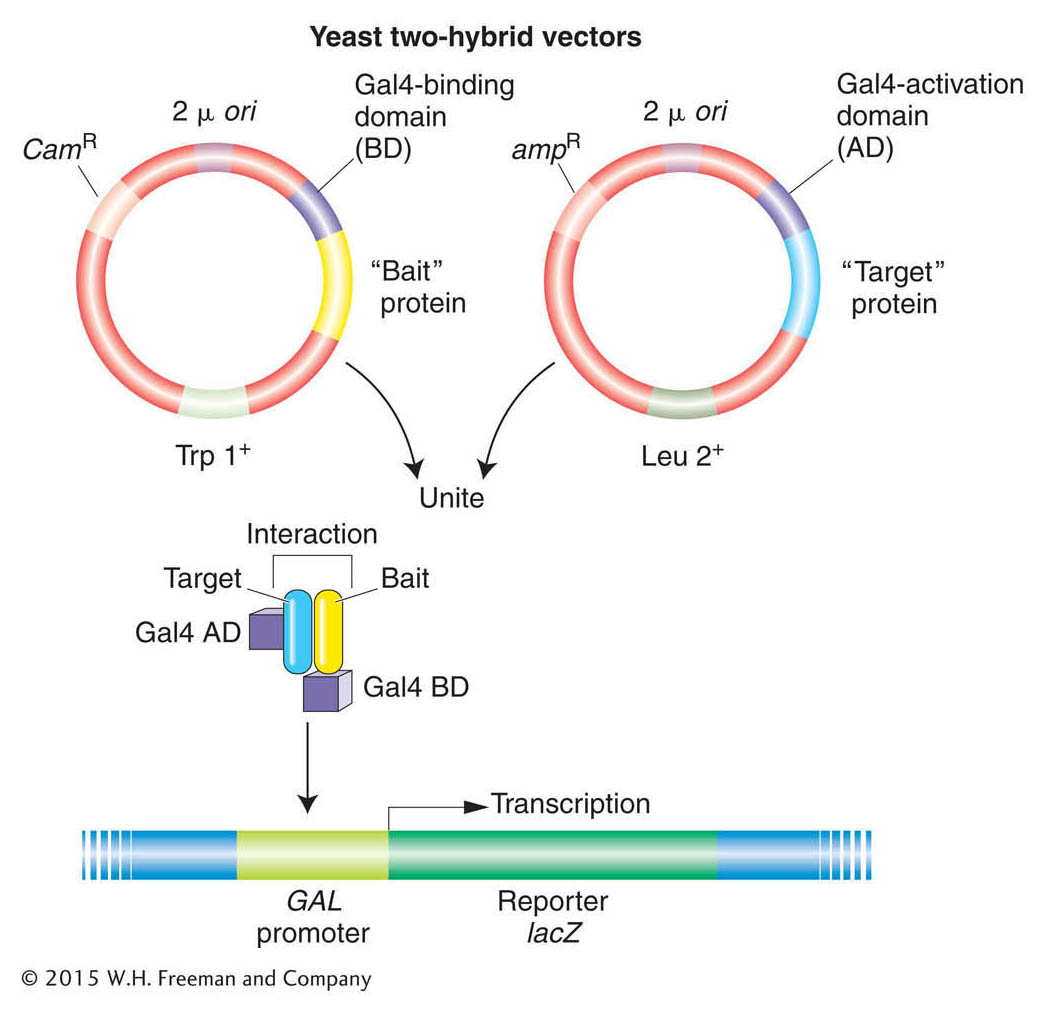
Figure 14-21: Studying protein interactions with the use of the yeast two-hybrid system
Figure 14-21: The system uses the binding of two proteins, a “bait” protein and a “target” protein, to restore the function of the Gal4 protein, which activates a reporter gene. Cam, Trp, and Leu are components of the selection systems for moving the plasmids around between cells. The reporter gene is lacZ, which resides on a yeast chromosome (shown in blue).
Studying the protein–DNA interactome using chromatin immunoprecipitation assay (ChIP) The sequence-specific binding of proteins to DNA is critical for correct gene expression. For example, regulatory proteins bind to promoters and activate or repress transcription in both bacteria and eukaryotes (see Chapters 11, 12, and 13). In the case of eukaryotes, chromosomes are organized into chromatin, in which the fundamental unit, the nucleosome, contains DNA wrapped around histones. Post-translational modification of histones often dictates what proteins bind and where (see Chapter 12). A variety of technologies have been developed that allow researchers to isolate specific regions of chromatin so that DNA and its associated proteins can be analyzed together. The most widely used method is called ChIP (chromatin immunoprecipitation), and its application is described below (Figure 14-22).
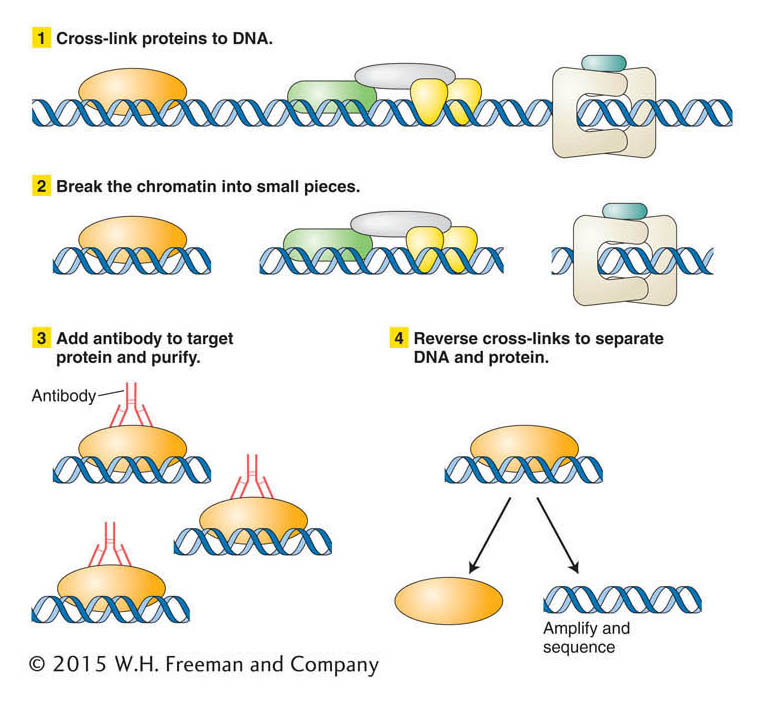
Figure 14-22: Steps in a chromatin immunoprecipitation assay (ChIP)
Figure 14-22: ChIP is a technique for isolating the DNA and its associated proteins in a specific region of chromatin so that both can be analyzed together.
Let’s say that you have isolated a gene from yeast and suspect that it encodes a protein that binds to DNA when yeast is grown at high temperature. You want to know whether this protein binds to DNA and, if so, to what yeast sequence. One way to address this question is first to treat yeast cells that have been grown at high temperature with a chemical that will cross-link proteins to the DNA. In this way proteins bound to the DNA at the time of chromatin isolation will remain bound through subsequent treatments. The next step is to break the chromatin into small pieces. To separate the fragment containing your protein–DNA complex from others, you use an antibody that reacts specifically with the encoded protein. You add your antibody to the mixture so that it forms an immune complex that can be purified. The DNA bound in the immune complex can be analyzed after cross-linking is reversed. DNA bound by the protein may be sequenced directly or amplified into many copies by PCR to prepare for DNA sequencing.
As you saw in Chapter 12, regulatory proteins often activate transcription of many genes simultaneously by binding to several promoter regions. A variation of the ChIP procedure, called ChIP-chip, has been devised to identify multiple binding sites in a sequenced genome. Proteins that bind to many genomic regions are immunoprecipitated as described above. Then, after cross-linking is reversed, the DNA fragments are labeled and used to probe microarray chips that contain the entire genomic sequence of the species under study.
Reverse genetics
The kinds of data obtained from microarray experiments and protein-interaction screens are suggestive of interactions within the genome and proteome, but they do not allow one to draw firm conclusions about gene functions and interactions in vivo. For example, finding out that the expression of certain genes is lost in some cancers is not proof of cause and effect. The gold standard for establishing the function of a gene or genetic element is to disrupt its function and to understand phenotypes in native conditions. Starting from available gene sequences, researchers can now use a variety of methods to disrupt the function of a specific gene. These methods are referred to as reverse genetics. Reverse-genetic analysis starts with a known molecule—a DNA sequence, an mRNA, or a protein—and then attempts to disrupt this molecule to assess the role of the normal gene product in the biology of the organism.
There are several approaches to reverse genetics and new technologies are constantly being developed and refined. One approach is to introduce random mutations into the genome but then to home in on the gene of interest by molecular identification of mutations in the gene. A second approach is to conduct a targeted mutagenesis that produces mutations directly in the gene of interest. A third approach is to create phenocopies—effects comparable to mutant phenotypes—usually by treatment with agents that interfere with the mRNA transcript of the gene.
Each approach has its advantages. Random mutagenesis is well established, but it requires that one sift through all the mutations to find those that include the gene of interest. Targeted mutagenesis can also be labor intensive but, after the targeted mutation has been obtained, its characterization is more straightforward. Creating phenocopies can be very efficient, especially as libraries of tools have been developed for particular model species. We will consider examples of each of these approaches.
Reverse genetics through random mutagenesis Random mutagenesis for reverse genetics employs the same kinds of general mutagens that are used for forward genetics: chemical agents, radiation, or transposable genetic elements. However, instead of screening the genome at large for mutations that exert a particular phenotypic effect, reverse genetics focuses on the gene in question, which can be done in one of two general ways.
One approach is to focus on the map location of the gene. Only mutations falling in the region of the genome where the gene is located are retained for further detailed molecular analysis. Thus, in this approach, the recovered mutations must be mapped. One straightforward way is to cross a new mutant with a mutant containing a known deletion or mutation of the gene of interest. Symbolically, the pairing is new mutant/known mutant. Only the pairings that result in progeny with a mutant phenotype (showing lack of complementation) are saved for study.
In another approach, the gene of interest is identified in the mutagenized genome and checked for the presence of mutations. For example, if the mutagen causes small deletions, then, after PCR amplification of gene fragments, genes from the parental and mutagenized genomes can be compared, looking for a mutagenized genome in which the gene of interest is reduced in size. Similarly, transposable-element insertions into the gene of interest can be readily detected because they increase its size. With the ability to sequence whole genomes of model species rapidly and relatively cheaply, one can also search for single-base-pair substitutions. In these ways, a set of genomes containing random mutations can be effectively screened to identify the small fraction of mutations that are of interest to a researcher.
Reverse genetics by targeted mutagenesis For most of the twentieth century, researchers viewed the ability to direct mutations to a specific gene as the unattainable “holy grail” of genetics. However, now several such techniques are available. After a gene has been inactivated in an individual, geneticists can evaluate the phenotype exhibited for clues to the gene’s function. While the tools for targeted gene mutations were first developed using genetic techniques for model organisms, new technologies are enabling the disruption and manipulation of genes in nonmodel species.
Gene-specific mutagenesis usually requires the replacement of a resident wild-type copy of an entire gene by a mutated version of that gene. The mutated gene inserts into the chromosome by a mechanism resembling homologous recombination, replacing the normal sequence with the mutant (Figure 14-23). This approach can be used for targeted gene knockout, in which a null allele replaces the wild-type copy. Some techniques are so efficient that, in E. coli and S. cerevisiae, for example, it has been possible to mutate every gene in the genome to try to ascertain its biological function.
KEY CONCEPT
Targeted mutagenesis is the most precise means of obtaining mutations in a specific gene and can now be practiced in a variety of model systems, including mice and flies.
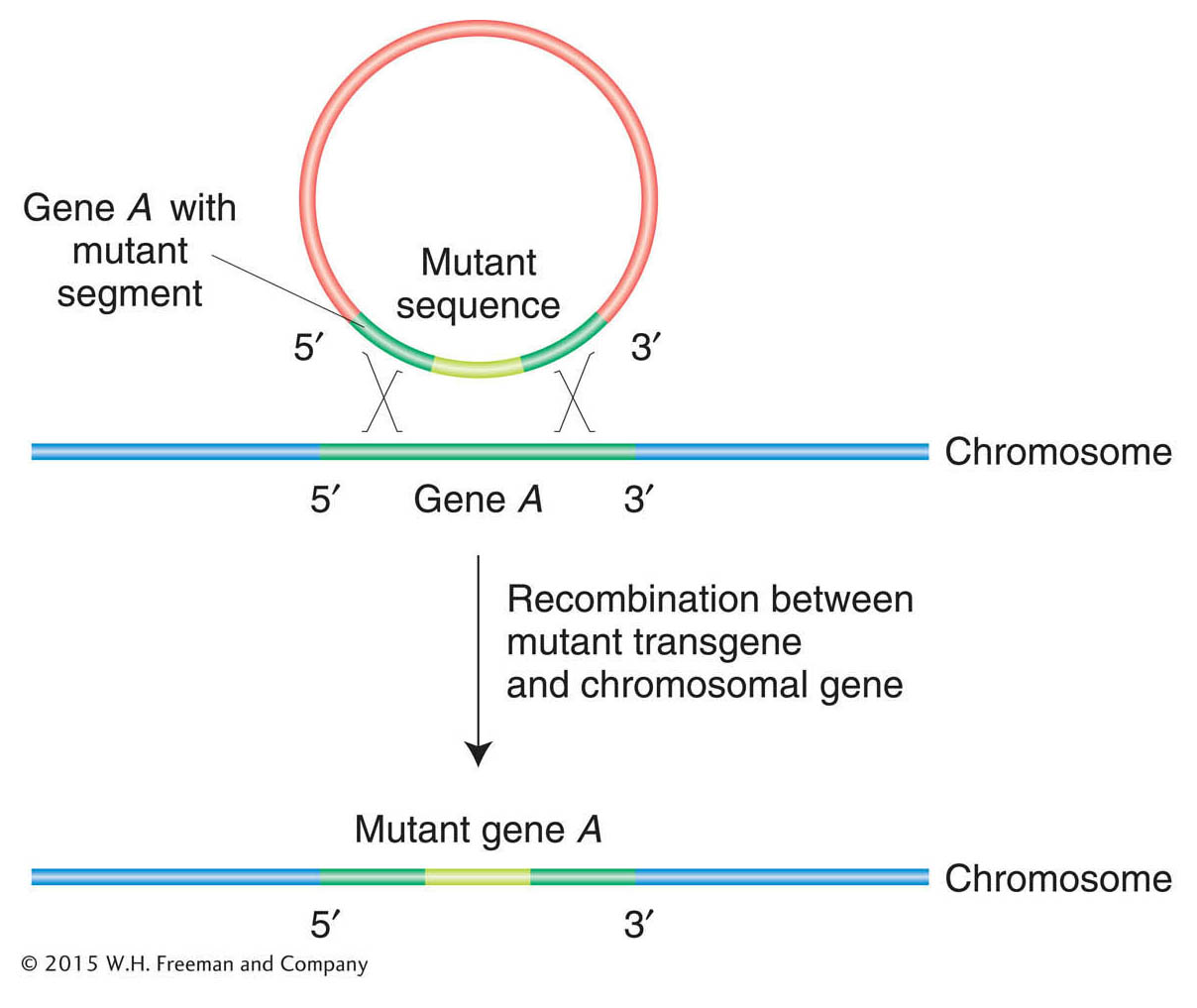
Figure 14-23: Disrupting gene function with the use of targeted mutagenesis
Figure 14-23: The basic molecular event in targeted gene replacement. A transgene containing sequences from two ends of a gene but with a selectable segment of DNA in between is introduced into a cell. Double recombination between the transgene and a normal chromosomal gene produces a recombinant chromosomal gene that has incorporated the abnormal segment.
Reverse genetics by phenocopying The advantage of inactivating a gene itself is that mutations will be passed on from one generation to the next, and so, once obtained, a line of mutants is always available for future study. On the other hand, phenocopying can be applied to a great many organisms regardless of how well developed the genetic technology is for a given species.
One of the most exciting discoveries of the past decade or so has been the discovery of a widespread mechanism whose natural function seems to be to protect a cell from foreign DNA. This mechanism is called RNA interference (RNAi). Researchers have capitalized on this cellular mechanism to make a powerful method for inactivating specific genes. The inactivation is achieved as follows. A double-stranded RNA is made with sequences homologous to part of the gene under study and is introduced into a cell (Figure 14-24). The RNA-induced silencing complex, or RISC, then degrades native mRNA that is complementary to the double-stranded RNA. The net result is a complete or considerable reduction of mRNA levels that lasts for hours or days, thereby nullifying expression of that gene. The technique has been widely applied in model systems such as C. elegans, Drosophila, zebrafish, and several plant species.
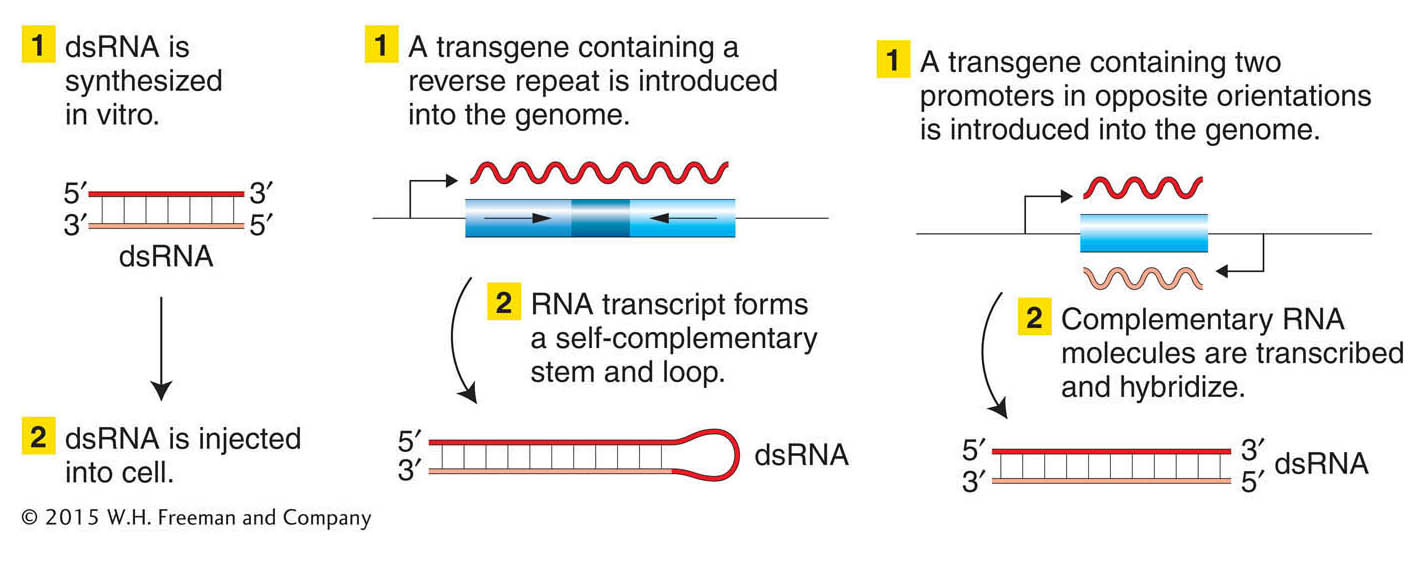
Figure 14-24: Disrupting gene function with the use of RNA interference
Figure 14-24: Three ways to create and introduce double-stranded RNA (dsRNA) into a cell. The dsRNA will then stimulate RNAi, degrading sequences that match those in the dsRNA.
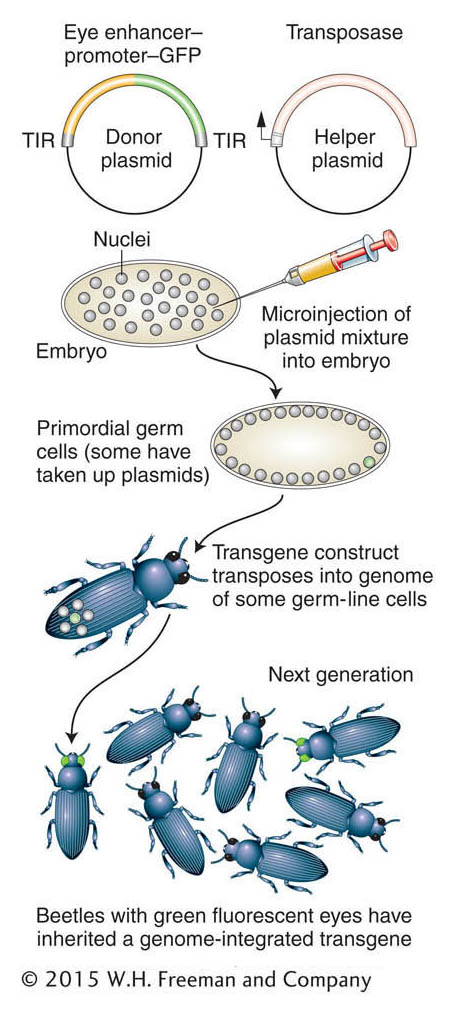
Figure 14-25: Inserting transgenes into a nonmodel organism
Figure 14-25: Creation of transgenic beetles expressing a green fluorescent protein. TIR, terminal inverted repeat.
But what makes RNAi especially powerful is that it can be applied to non-model organisms. First, target genes of interest can be identified by comparative genomics. Then RNAi sequences are produced to target the inhibition of the specific target genes. This technique has been applied, for example, to a mosquito that carries malaria (Anopheles gambiae). Using these techniques, scientists can better understand the biological mechanisms relating to the medical or economic effect of such species. The genes that control the complicated life cycle of the malaria parasite, partly inside a mosquito host and partly inside the human body, can be better understood, revealing new ways to control the single most common infectious disease in the world.
KEY CONCEPT
RNAi-based methods provide general ways of experimentally interfering with the function of a specific gene without changing its DNA sequence (generally called phenocopying).Functional genomics with nonmodel organisms Much of our consideration of mutational dissection and phenocopying has focused on genetic model organisms. One current focus of many geneticists is the broader application of these techniques, including to species that have negative effects on human society, such as parasites, disease carriers, or agricultural pests. Classical genetic techniques are not readily applicable to most of these species, but the roles of specific genes can be assessed through the insertion of transgenes and the generation of phenocopies.
The insertion of transgenes into a beetle is shown in Figure 14-25. Transgenic beetles can be produced by using methodology similar to that used to produce transgenic Drosophila (see Chapter 10). However, some way is needed to identify successful transgenesis. Therefore, the technique depends on using a reporter gene that can be expressed in a wild-type recipient. The green fluorescent protein (GFP), originally isolated from a jellyfish, is a useful reporter for this application. As in Drosophila, transgenes are inserted as parts of transposons, and a helper plasmid encoding a transposase facilitates insertion of the transposon bearing the transgene. Figure 14-25 shows the use of GFP transgenes driven by an enhancer element that drives expression in the insect eye. This method has also been effectively used to create GFP-expressing transgenes in the mosquito species that carries yellow fever and dengue fever (Aedes aegypti), a flour beetle (Tribolium castaneum), and the silkworm moth (Bombyx mori) (Figure 14-26). Often, the GFP transgene is used simply as a genetic marker for experiments in which an RNAi construct or other transgene is co-inserted in order to manipulate gene function.

Figure 14-26: Examples of nonmodel insects expressing a transgene
Figure 14-26: Examples of a transgenic green fluorescent protein reporter expressed in the eyes of some nonmodel insects. Expression is driven from one single promoter active in the eye. The insects are mosquito (Aedes aegypti, top) and silkworm moth (Bombyx mori, bottom).
[(Top) AP Photo/Jacquelyn Martin; (bottom) Marek Jindra.]