16.2 The Molecular Basis of Spontaneous Mutations
Gene mutations can arise spontaneously or they can be induced. Spontaneous mutations are naturally occurring mutations and arise in all cells. Induced mutations arise through the action of certain agents called mutagens that increase the rate at which mutations occur. In this section, we consider the nature of spontaneous mutations.
Luria and Delbrück fluctuation test
The origin of spontaneous hereditary change has always been a topic of considerable interest. Among the first questions asked by geneticists was, Do spontaneous mutations occur in response to the selecting agent or are variants present at a low frequency in most populations? An ideal experimental system to address this important question was the analysis of mutations in bacteria that confer resistance to specific environmental agents not normally tolerated by wild-
One experiment by Salvador Luria and Max Delbrück in 1943 was particularly influential in shaping our understanding of the nature of mutation, not only in bacteria, but in organisms generally. It was known at the time that, if E. coli bacteria are spread on a plate of nutrient medium in the presence of phage T1, the phages soon infect and kill the bacteria. Rarely, but regularly, colonies were seen that were resistant to phage attack; these colonies were stable and so appeared to be genuine mutants. However, whether these mutants were produced spontaneously but randomly in time or the presence of the phage induced a physiological change that caused resistance was not known.
587
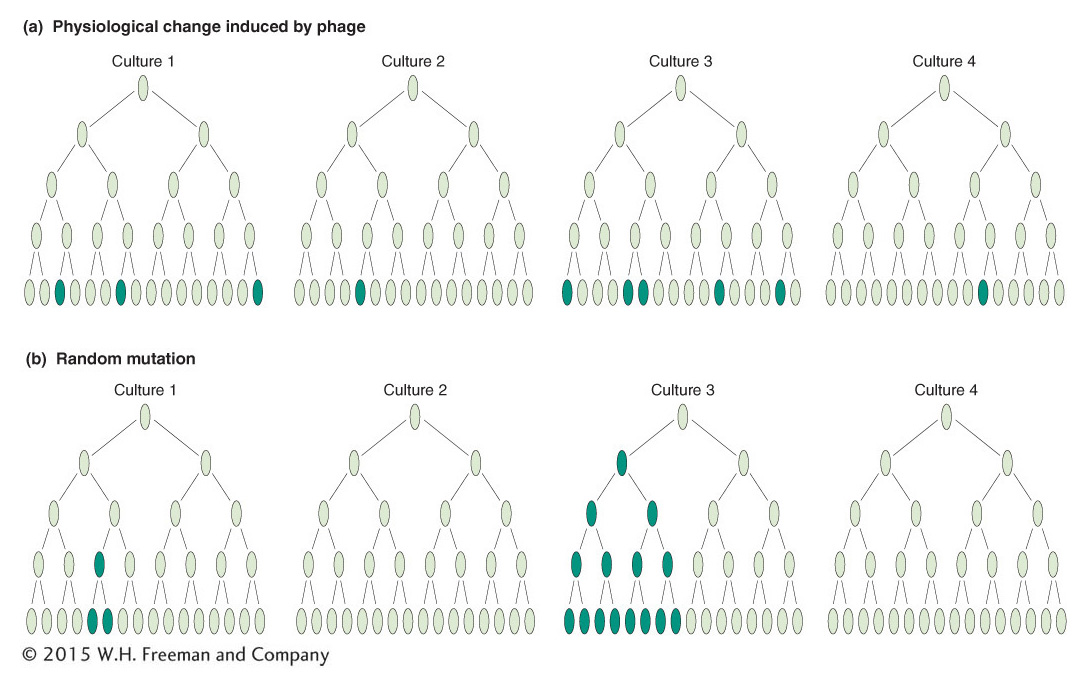
Luria reasoned that, if mutations occurred spontaneously, then the mutations might be expected to occur at different times in different cultures. In this case, the numbers of resistant colonies per culture should show high variation (or “fluctuation” in his word). He later claimed that the idea came to him as he watched the fluctuating returns obtained by colleagues gambling on a slot machine at a faculty dance in a local country club; hence the origin of the term “jackpot” mutation.
Luria and Delbrück designed their “fluctuation test” as follows. They inoculated 20 small cultures, each with a few cells, and incubated them until there were 108 cells per milliliter. At the same time, a much larger culture also was inoculated and incubated until there were 108 cells per milliliter. The 20 individual cultures and 20 samples of the same size from the large culture were plated in the presence of phage. The 20 individual cultures showed high variation in the number of resistant colonies: 11 plates had 0 resistant colonies, and the remainder had 1, 1, 3, 5, 5, 6, 35, 64, and 107 per plate (Figure 16-5a). The 20 samples from the large culture showed much less variation from plate to plate, all in the range of 14 to 26. If the phage were inducing mutations, there was no reason why fluctuation should be higher on the individual cultures because all were exposed to phage similarly. The best explanation was that mutation was occurring randomly in time: the early mutations gave the higher numbers of resistant cells because the mutant cells had time to produce many resistant descendants. The later mutations produced fewer resistant cells (Figure 16-5b). This result led to the reigning “paradigm” of mutation; that is, whether in viruses, bacteria, or eukaryotes, mutations can occur in any cell at any time and their occurrence is random. For this and other work, Luria and Delbrück were awarded the Nobel Prize in Physiology or Medicine in 1969. Interestingly, this was after Luria’s first graduate student, James Watson, won his Nobel Prize (with Francis Crick in 1964) for the discovery of the DNA double-
588
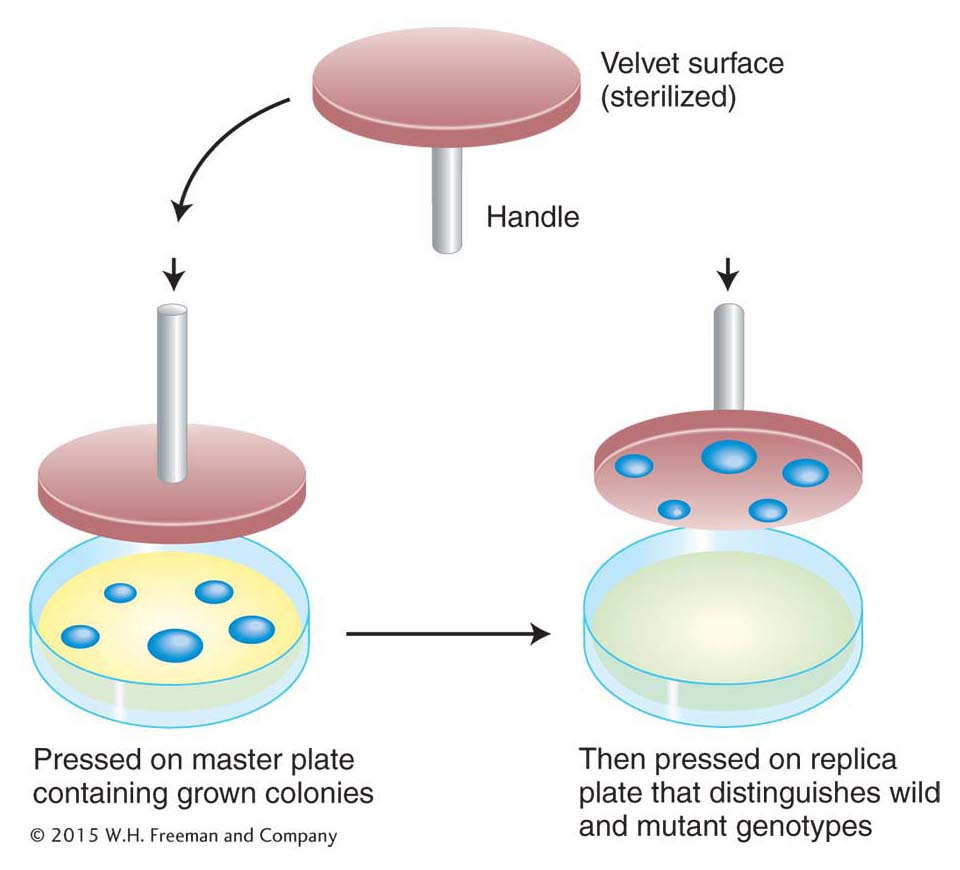
This elegant analysis suggests that the resistant cells are selected by the environmental agent (here, phage) rather than produced by it. Can the existence of mutants in a population before selection be demonstrated directly? This demonstration was made possible by the use of a technique called replica plating, developed largely by Esther Lederberg in 1952. A population of bacteria was plated on nonselective medium—
KEY CONCEPT
Mutation is a random process. Any allele in any cell may mutate at any time.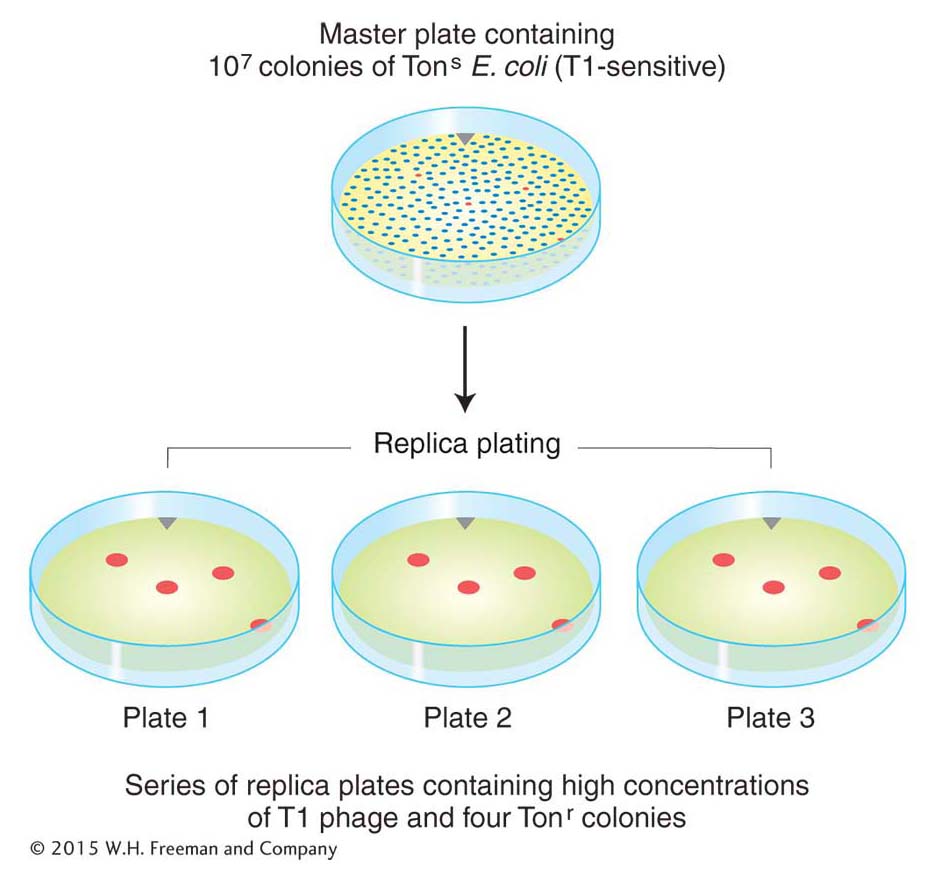
Mechanisms of spontaneous mutations
Spontaneous mutations arise from a variety of sources. One source is the DNA-
Errors in DNA replication An error in DNA replication can result when an illegitimate nucleotide pair (say, A–
Transitions You saw in Chapter 7 that each of the bases in DNA can appear in one of several tautomeric forms that can pair to the wrong base. Mismatches can also result when one of the bases becomes ionized. This type of mismatch may occur more frequently than mismatches due to tautomerization. These errors are frequently corrected by the proofreading (editing) function of bacterial DNA pol III (see Figure 7-18). If proofreading does not occur, all the mismatches described so far lead to transition mutations, in which a purine substitutes for a purine or a pyrimidine for a pyrimidine (see Figure 16-2). Other repair systems (described later in this chapter) correct many of the mismatched bases that escape correction by the polymerase editing function.
589
Transversions In transversion mutations, a pyrimidine substitutes for a purine, or vice versa (see Figure 16-2). The creation of a transversion by a replication error would require, at some point in the course of replication, the mispairing of a purine with a purine or a pyrimidine with a pyrimidine. Although the dimensions of the DNA double helix render such mismatches energetically unfavorable, we now know from X-
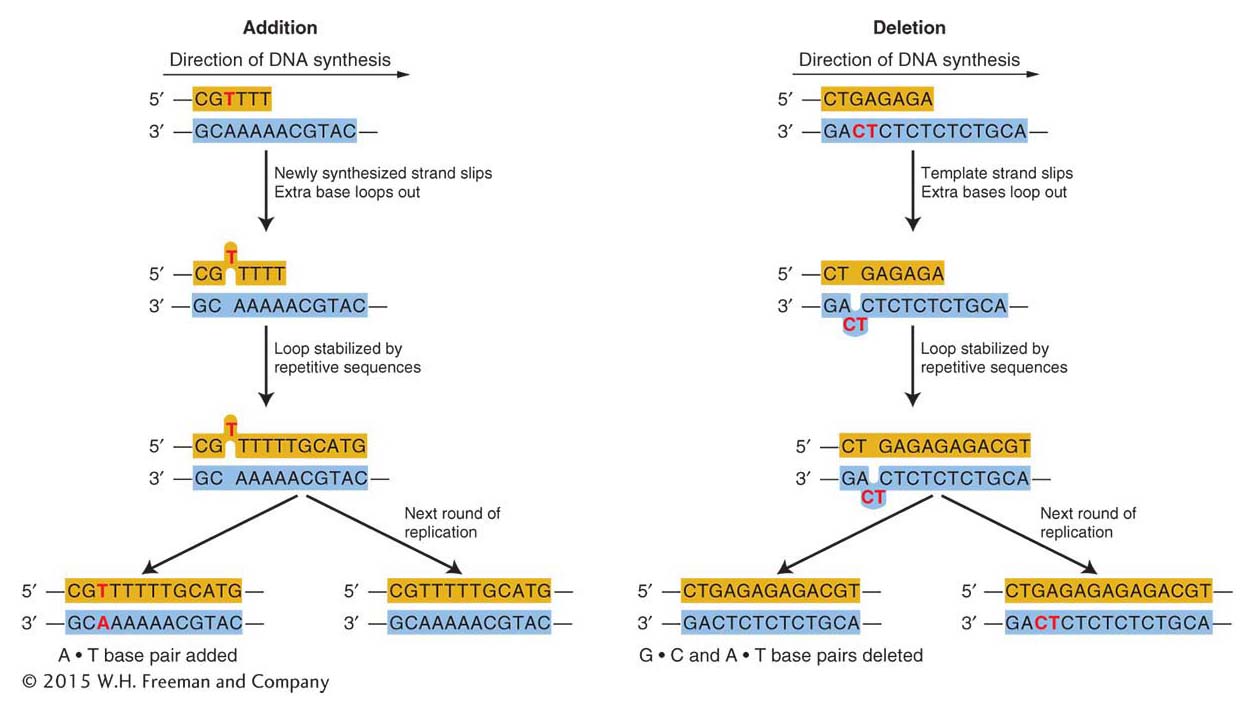
Frameshift mutations Replication errors can also lead to frameshift mutations. Recall from Chapter 9 that such mutations result in greatly altered proteins.
Certain kinds of replication errors can lead to indel mutations—that is, insertions or deletions of one or more base pairs. These insertions or deletions produce frameshift mutations when they add or subtract a number of bases not divisible by three (the size of a codon) in the protein-
ANIMATED ART: Molecular mechanism of mutation
Spontaneous lesions In addition to replication errors, spontaneous lesions, naturally occurring damage to the DNA, can generate mutations. Two of the most frequent spontaneous lesions result from depurination and deamination.
Depurination, the more common of the two, is the loss of a purine base. Depurination consists of the interruption of the glycosidic bond between the base and deoxyribose and the subsequent loss of a guanine or an adenine residue from the DNA. The DNA backbone remains intact.
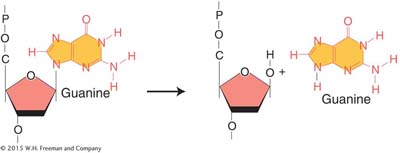
A mammalian cell spontaneously loses about 10,000 purines from its DNA in a 20-
590
The deamination of cytosine yields uracil.
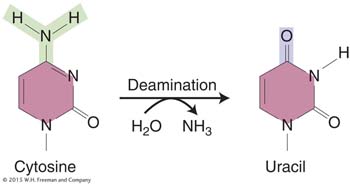
Unrepaired uracil residues will pair with adenine in replication, resulting in the conversion of a G · C pair into an A · T pair (a G · C → A · T transition).
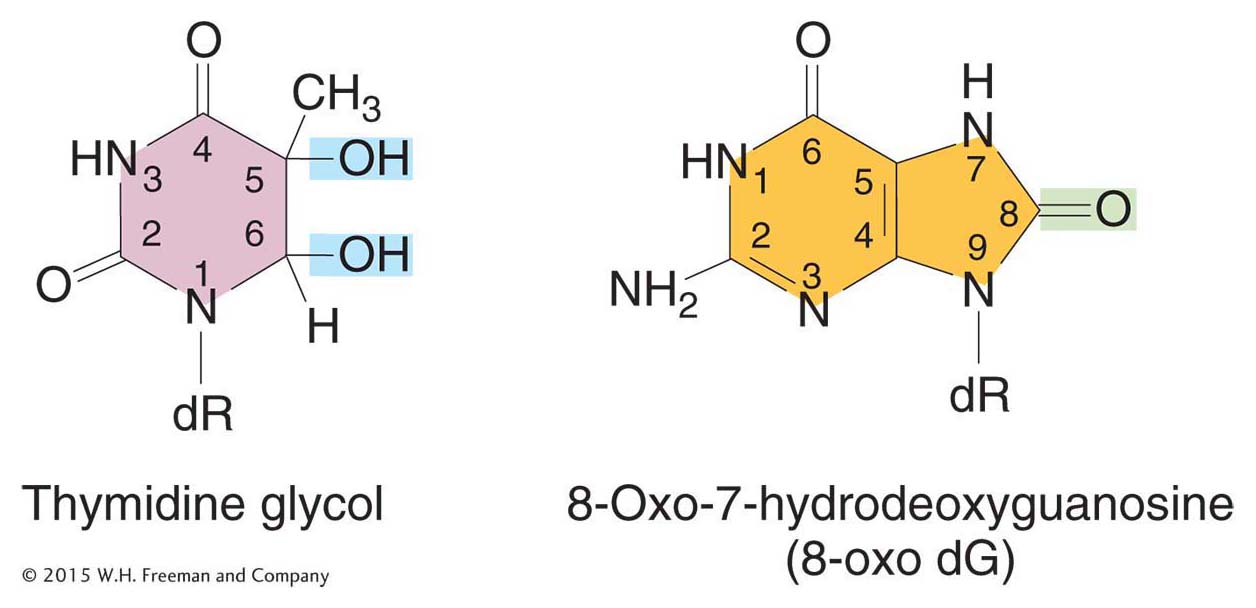
Oxidatively damaged bases represent a third type of spontaneous lesion that generates mutations. Active oxygen species, such as superoxide radicals (O2 · –), hydrogen peroxide (H2O2), and hydroxyl radicals (· OH), are produced as by-
KEY CONCEPT
Spontaneous mutations can be generated by different processes. Replication errors and spontaneous lesions generate most spontaneous base substitutions. Replication errors can also cause deletions that lead to frameshift mutations.591
Spontaneous mutations in humans: trinucleotide-repeat diseases
DNA sequence analysis has revealed the gene mutations contributing to numerous human hereditary diseases. Many are of the expected base-
A common mechanism responsible for a number of genetic diseases is the expansion of a three-
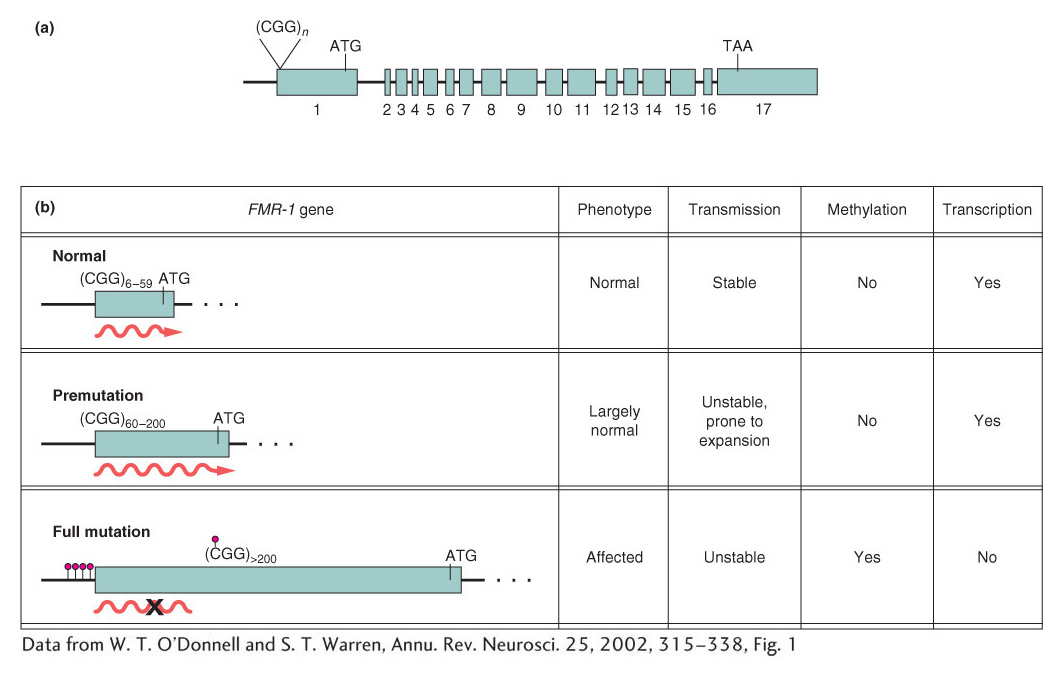
How does repeat number correlate with the disease phenotype? Humans normally show considerable variation in the number of CGG repeats in the FMR-
592
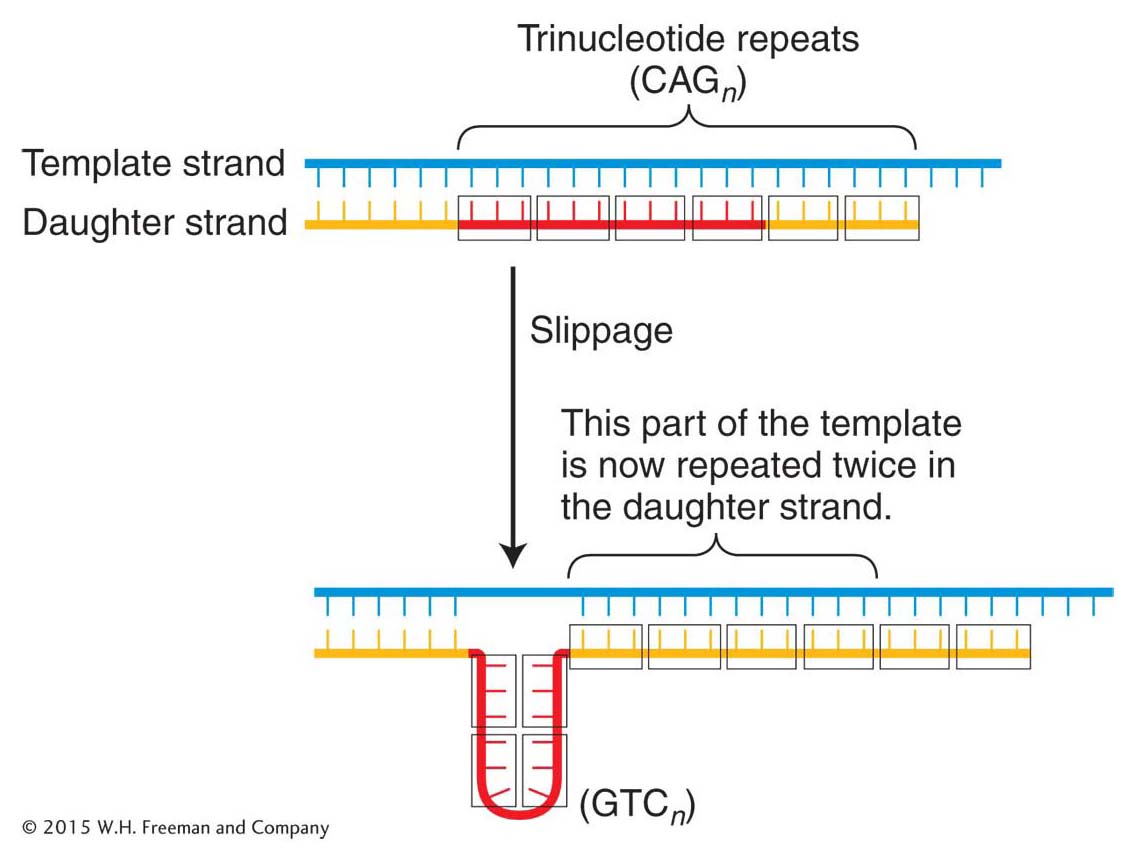
The proposed mechanism for the generation of these repeats is replication slippage that occurs in the course of DNA synthesis (Figure 16-11). However, the extraordinarily high frequency of mutation at the trinucleotide repeats in fragile X syndrome suggests that in human cells, after a threshold level of about 50 repeats, the replication machinery cannot faithfully replicate the correct sequence and large variations in repeat numbers result.
Other diseases, such as Huntington disease (see Chapter 2), also have been associated with the expansion of trinucleotide repeats in a gene or its regulatory regions. Several general themes apply to these diseases. In Huntington disease, for example, the wild-
Huntington disease and Kennedy disease (also called X-
Properties common to some trinucleotide-
However, this explanation cannot hold for all trinucleotide-
KEY CONCEPT
Trinucleotide-593