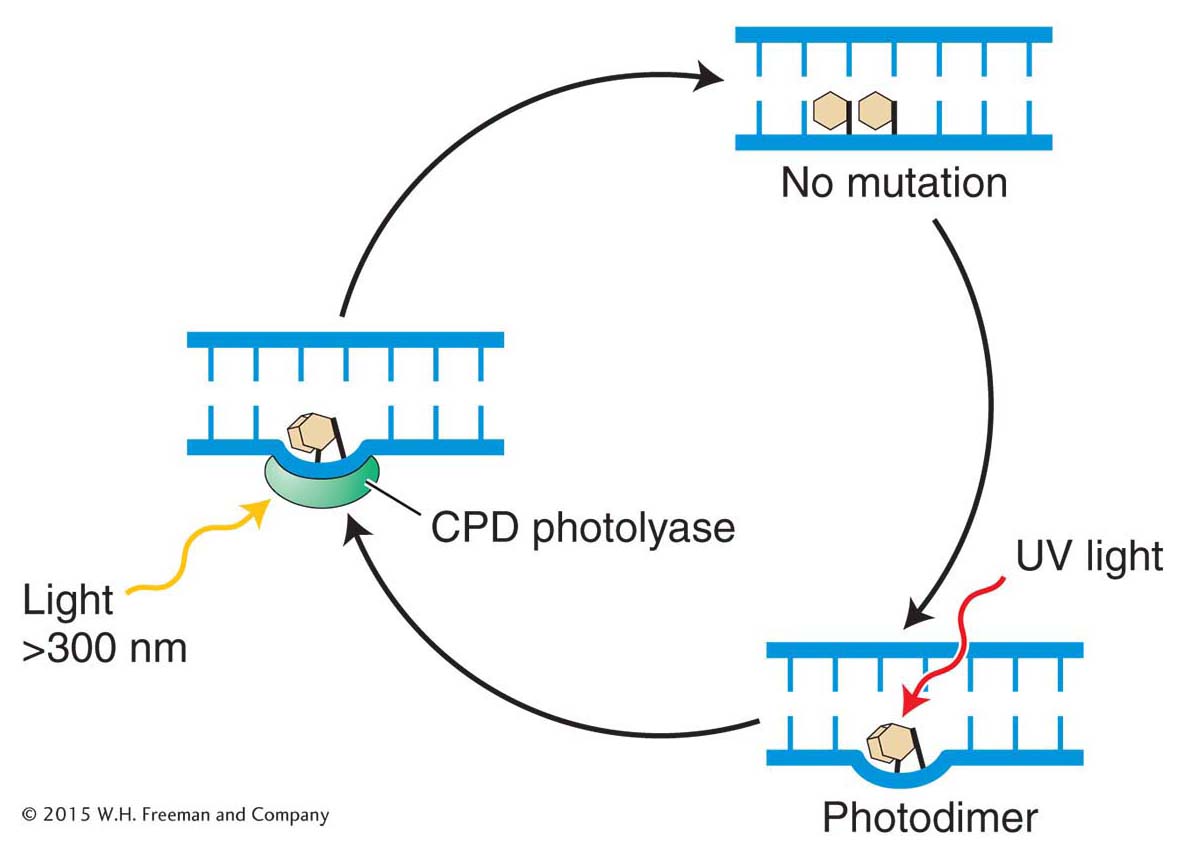
Base-excision repair
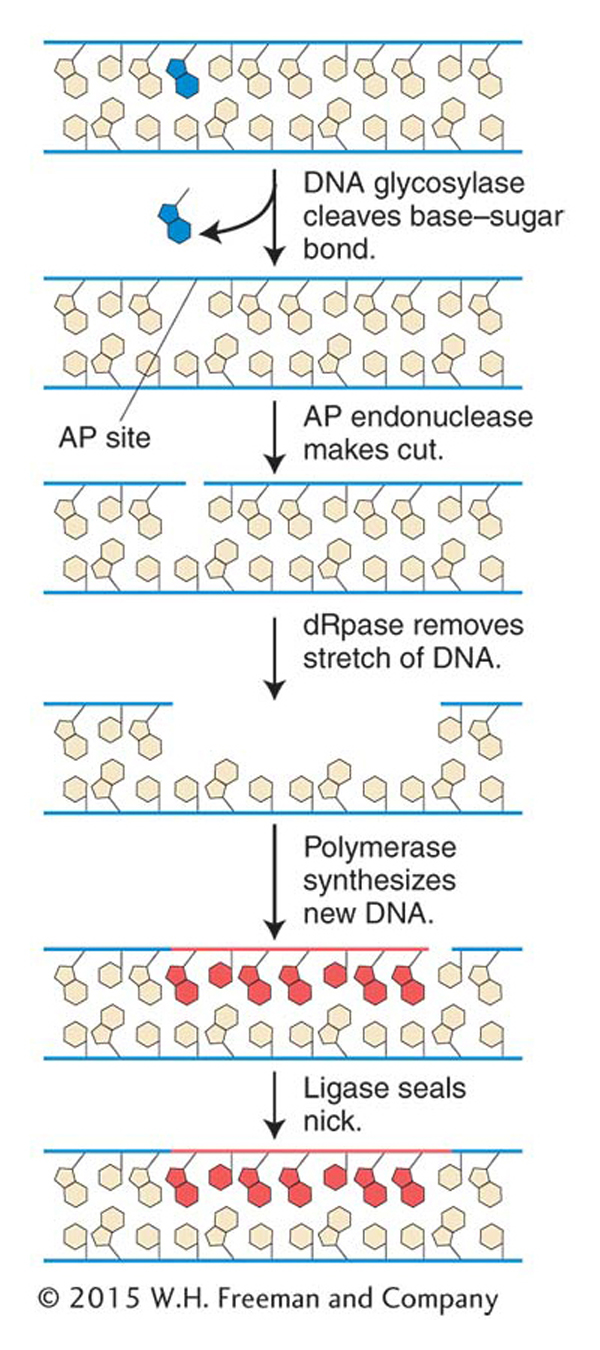
Figure 16-20: Minor base damage is detected and repaired by base-excision repair
Figure 16-20: In base-excision repair, damaged bases are removed and repaired through sequential action of a DNA glycosylase, AP endonuclease, deoxyribophos-phodiesterase (dRpase), DNA polymerase, and ligase.
An overarching principle guiding cellular genetic systems is the power of nucleotide sequence complementarity. (Recall that genetic analysis also depends heavily on this principle.) Important repair systems exploit the properties of antiparallel complementarity to restore damaged DNA segments to their initial, undamaged state. In these systems, a base or longer segment of a DNA chain is removed and replaced with a newly synthesized nucleotide segment complementary to the opposite template strand. Unlike the examples of reversal of damage described in the preceding section, these pathways include the removal and replacement of one or more bases.
The first repair system of this type that we will examine is base-excision repair. After DNA proofreading by DNA polymerase, base-excision repair is the most important mechanism used to remove incorrect or damaged bases. The main target of base-excision repair is nonbulky damage to bases. This type of damage can result from the variety of causes mentioned in preceding sections, including methylation, deamination, oxidation, or the spontaneous loss of a DNA base. Base-excision repair (Figure 16-20) is carried out by DNA glycosylases that cleave base–sugar bonds, thereby liberating the altered bases and generating apurinic or apyrimidinic (AP) sites. An enzyme called AP endonuclease then nicks the damaged strand upstream of the AP site. A third enzyme, deoxyribophospho-diesterase, cleans up the backbone by removing a stretch of neighboring sugar–phosphate residues so that a DNA polymerase can fill the gap with nucleotides complementary to the other strand. DNA ligase then seals the new nucleotide into the backbone (see Figure 16-20).
Numerous DNA glycosylases exist. One, uracil-DNA glycosylase, removes uracil from DNA. Uracil residues, which result from the spontaneous deamination of cytosine, can lead to a C-to-T transition if unrepaired. One advantage of having thymine (5-methyluracil) rather than uracil as the natural pairing partner of adenine in DNA is that spontaneous cytosine deamination events can be recognized as abnormal and then excised and repaired. If uracil were a normal constituent of DNA, such repair would not be possible.
However, deamination does pose other problems for both bacteria and eukaryotes. By analyzing a large number of mutations in the lacI gene, Jeffrey Miller identified places in the gene where one or more bases were prone to frequent mutation. Miller found that these so-called mutational hotspots corresponded to deaminations at certain cytosine residues. DNA sequence analysis of G · C → T · A transition hot spots in the lacI gene showed that 5-methylcytosine residues are present at each hotspot. Recall from Chapter 12 that eukaryotic DNA may be methylated to inactive genes. Similarly, E. coli and other bacteria also methylate their DNA, although for different purposes. Some of the data from this lacI study are shown in Figure 16-21. The height of each bar on the graph represents the frequency of mutations at each site. The positions of 5-methylcytosine residues can be seen to correlate nicely with the most mutable sites.
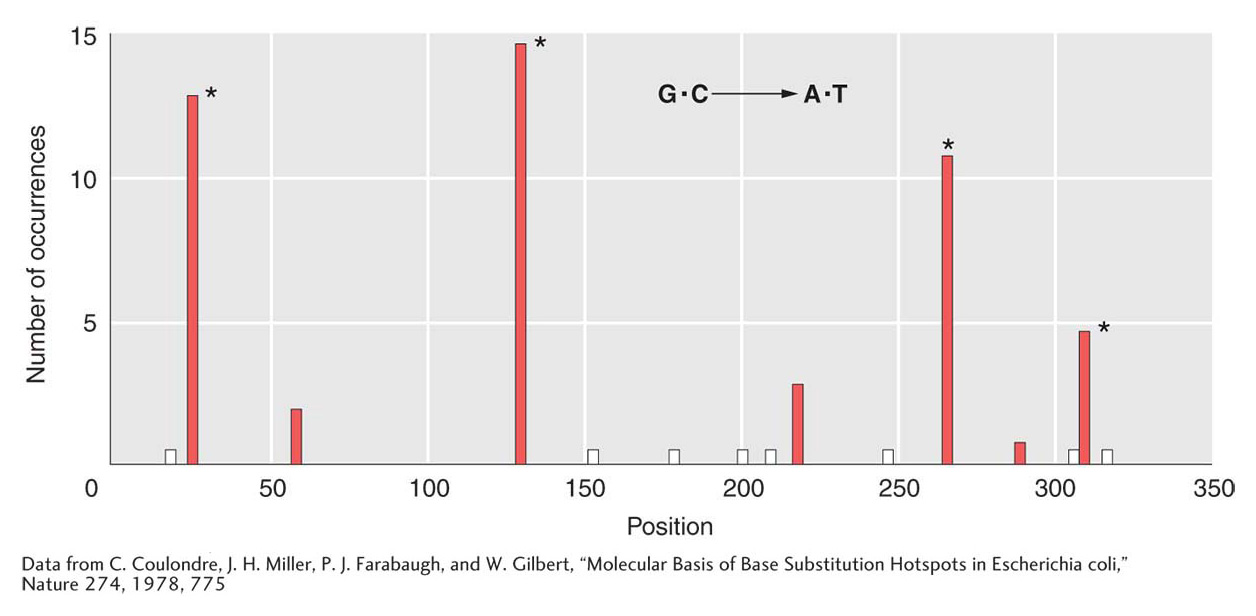
Figure 16-21: 5-Methylcytosine is a hotspot for mutation
Figure 16-21: Methylcytosine hotspots in E. coli. Nonsense mutations at 15 different sites in lacl were scored. All resulted in G · C → A · T transitions. The asterisk (*) marks the positions of 5-methylcytosines, and the white bars mark sites where transitions known to occur were not isolated in this group.
[Data from C. Coulondre, J. H. Miller, P. J. Farabaugh, and W. Gilbert, “Molecular Basis of Base Substitution Hotspots in Escherichia coli,” Nature 274, 1978, 775.]
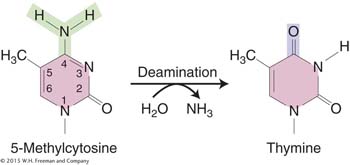
Why are 5-methylcytosines hot spots for mutations? The deamination of 5-methylcytosine generates thymine (5-methyluracil):
Thymine is not recognized by the enzyme uracil-DNA glycosylase and thus is not repaired. Therefore, C → T transitions generated by deamination are seen more frequently at 5-methylcytosine sites because they escape this repair system. A consequence of the frequent mutation of 5-methylcytosine to thymine is that methylated regions of the genome (which are usually transcriptionally inactive; see Chapter 12) are converted, over evolutionary time, to AT-rich regions. In contrast, coding and regulatory regions, which are less methylated, remain GC rich.
KEY CONCEPT
In base-excision repair, nonbulky damage to the DNA is recognized by one of several enzymes called DNA glycosylases that cleave the base-sugar bonds, releasing the incorrect base. Repair consists of the removal of the site that now lacks a base and the insertion of the correct base as guided by the complementary base in the undamaged strand.
Nucleotide-excision repair
Although the vast majority of the damage sustained by an organism is minor base damage that can be handled by base-excision repair, this mechanism can neither correct bulky adducts that distort the DNA helix, adducts such as the cyclobutane pyrimidine dimers caused by UV light (see Figure 16-15), nor correct damage to more than one base. A DNA polymerase cannot continue DNA synthesis past such lesions, and so the result is a replication block. A blocked replication fork can cause cell death. Similarly, an abnormal or damaged base can stall the transcription complex. To cope with both of these situations, prokaryotes and eukaryotes utilize an extremely versatile pathway called nucleotide-excision repair (NER) that is able to relieve replication and transcription blocks and repair the damage.
Interestingly, two autosomal recessive diseases in humans, xeroderma pigmentosum (XP) and Cockayne syndrome, are caused by defects in nucleotide-excision repair. Although patients with either XP or Cockayne syndrome are exceptionally sensitive to UV light, other symptoms are dramatically different. Xeroderma pigmentosum was introduced at the beginning of this chapter and is characterized by the early development of cancers, especially skin cancer and, in some cases, neurological defects. In contrast, patients afflicted with Cockayne syndrome have a variety of developmental disorders including dwarfism, deafness, and retardation. In broad terms, XP patients get early cancer, whereas Cockayne syndrome patients age prematurely. How can defects in the same repair pathway lead to such different disease symptoms? Although there is no simple answer to this question, work on the genetic basis of these diseases has led to the identification of important proteins in the NER pathway.
Nucleotide-excision repair is a complex process that requires dozens of proteins. Despite this complexity, the repair process can be divided into four phases:
Recognition of damaged base(s)
Assembly of a multiprotein complex at the site
Cutting of the damaged strand several nucleotides upstream and downstream of the damage site and removal of the nucleotides (~30) between the cuts
Use of the undamaged strand as a template for DNA polymerase followed by strand ligation
The fact that, as already mentioned, both stalled replication forks and stalled transcription complexes activate this repair pathway implies that there are two types of nucleotide-excision repair that differ in damage recognition (step 1). We now know that one type, called global genomic nucleotide-excision repair (GG-NER) corrects lesions anywhere in the genome and is activated by stalled replication forks. The other type repairs transcribed regions of DNA and is called, not surprisingly, transcription-coupled nucleotide-excision repair (TC-NER). As can be seen in Figure 16-22, although the recognition step differs, both GG-NER and TC-NER share the last four steps.
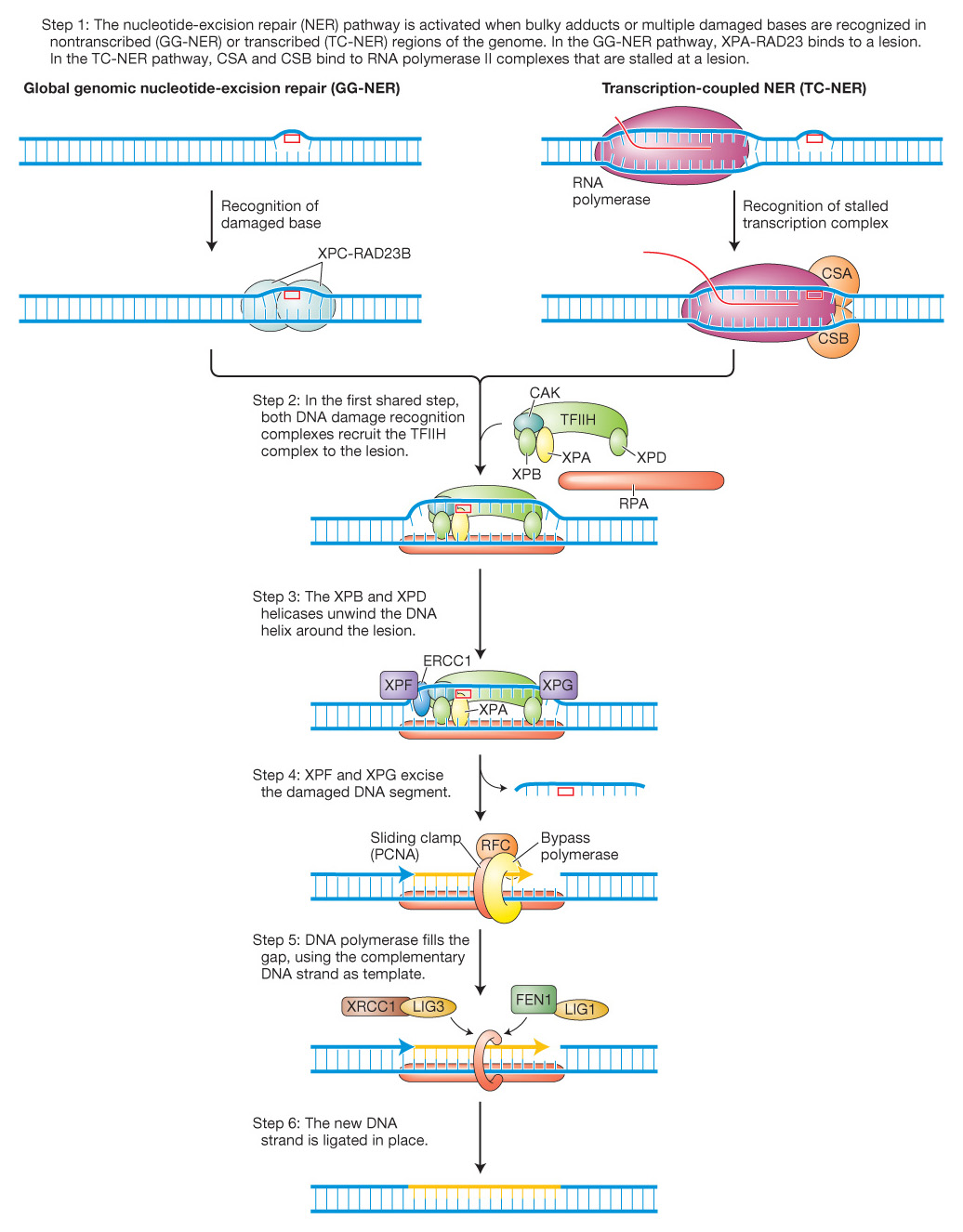
Figure 16-22: Two pathways for nucleotide-excision repair
Figure 16-22: The nucleotide-excision repair pathway is activated when bulky adducts or multiple damaged bases are recognized in nontranscribed (GG-NER) or transcribed (TC-NER) regions of the genome. These two pathways are initiated by different events and distinct complexes, as shown in the first step. Both complexes serve to attract the same TFIIH complex (Step 2). The recognition complexes are removed in Step 3 (not shown). In steps 4-6, a multiprotein complex excises several bases and resynthesizes them using the opposite strand as a template. See text for details.
At this point, you might be asking yourself, What if differences in the disease symptoms of XP and Cockayne syndrome were due to mutations in different classes of recognition proteins? You would be on the right track in asking that question. Patients with XP fall into 8 complementation groups, carrying mutations in one of 8 genes encoding proteins XPA through XPG (Figure 16-22). Patients with Cockayne syndrome have a mutation in one of two proteins called CSA and CSB, which are thought to recognize stalled transcription complexes.
GG-NER is initiated when a protein complex of XPC and RAD23B recognizes a distorted double helix caused by a damaged base and binds to the opposite strand. In contrast, TC-NER is initiated when an RNA polymerase complex is stalled by a DNA lesion in the transcribed strand and CSA and CSB bind at this site to form a recognition complex. After lesion recognition, the GG-NER and TC-NER pathways utilize largely the same proteins to remove and repair the damaged DNA because the role of both XPC-RAD23B and CSA/CSB is to attract the multiprotein TFIIH complex. Two of its 10 subunits, XPB and XPD, are helicases (3′-to-5′ and 5′-to-3′, respectively) that unwind and open the DNA helix around the lesion. Subsequent steps common to GG-NER and TC-NER mediate the cleavage and excision of the damaged base and as many as 30 adjacent nucleotides followed by DNA synthesis to fill the gap (see details in Figure 16-22). In addition to XPC-RAD23D, XPB, and XPD, XP patients harbor mutations in other proteins that participate in the common steps of NER. As shown in Figure 16-22, XPA promotes the release of the CAK subunit and the binding of RPA while endonucleases XPF (with ERCCI) and XPG cut 5′ and 3′, respectively, of the DNA damage. After removal of the damaged base and surrounding DNA, the gap is filled by a DNA polymerase assisted by the RFC and PCNA proteins. The last step of NER involves ligation of the new strand to the surrounding DNA by one of two ligation complexes (XRCC1/LIG3 or FEN1/LIG1).
Can the molecular differences between GG-NER and TC-NER provide an explanation for the different symptoms displayed by patients with XP and Cockayne syndrome? Recall that XP patients develop early cancers, whereas Cockayne syndrome patients have a variety of symptoms associated with premature aging. We have seen that the repair system of Cockayne syndrome patients cannot recognize stalled transcription complexes. A consequence of this defect is that the cell is more likely to activate the apoptosis suicide pathway. In a healthy person, cell death is often preferable to the propagation of a cell that has sustained DNA damage. However, according to this theory, the cell-death pathway would be activated more frequently in a Cockayne syndrome patient, thus leading to a variety of premature-aging symptoms. In contrast, XP patients can recognize stalled transcription complexes (they have normal CSA and CSB proteins) and prevent cell death when transcription is restarted. However, they cannot repair the original damage because of mutations in one of their XP proteins. Thus, mutations will accumulate in the cells of patients with XP and, as stated earlier in this chapter, the presence of mutations, whether caused by mutagens or the failure of repair pathways, increases the risk of developing many types of cancer.
KEY CONCEPT
Nucleotide-excision repair is a versatile pathway that recognizes and corrects DNA lesions due largely to UV damage and, in doing so, relieves stalled replication forks and transcription complexes. Patients with xeroderma pigmentosum and Cockayne syndrome are UV sensitive owing to mutations in key nucleotide-excision-repair proteins that recognize or repair the damaged bases.
Postreplication repair: mismatch repair
You learned in the first half of this chapter that many errors occur in DNA replication. In fact, the error rate is about 10–5. Correction by the 3′-to-5′ proofreading function of the replicative polymerase reduces the error rate to less than 10–7. The major pathway that corrects the remaining replicative errors is called mismatch repair. This repair pathway reduces the error rate to less than 10–9 by recognizing and repairing mismatched bases and small loops caused by the insertion and deletion of nucleotides (indels) in the course of replication. From these values, you can see that mutations leading to the loss of the mismatch-repair pathway could increase the mutation frequency 100-fold. In fact, loss of mismatch repair is associated with hereditary forms of colon cancer.
Mismatch-repair systems have to do at least three things:
Recognize mismatched base pairs
Determine which base in the mismatch is the incorrect one
Excise the incorrect base and carry out repair synthesis
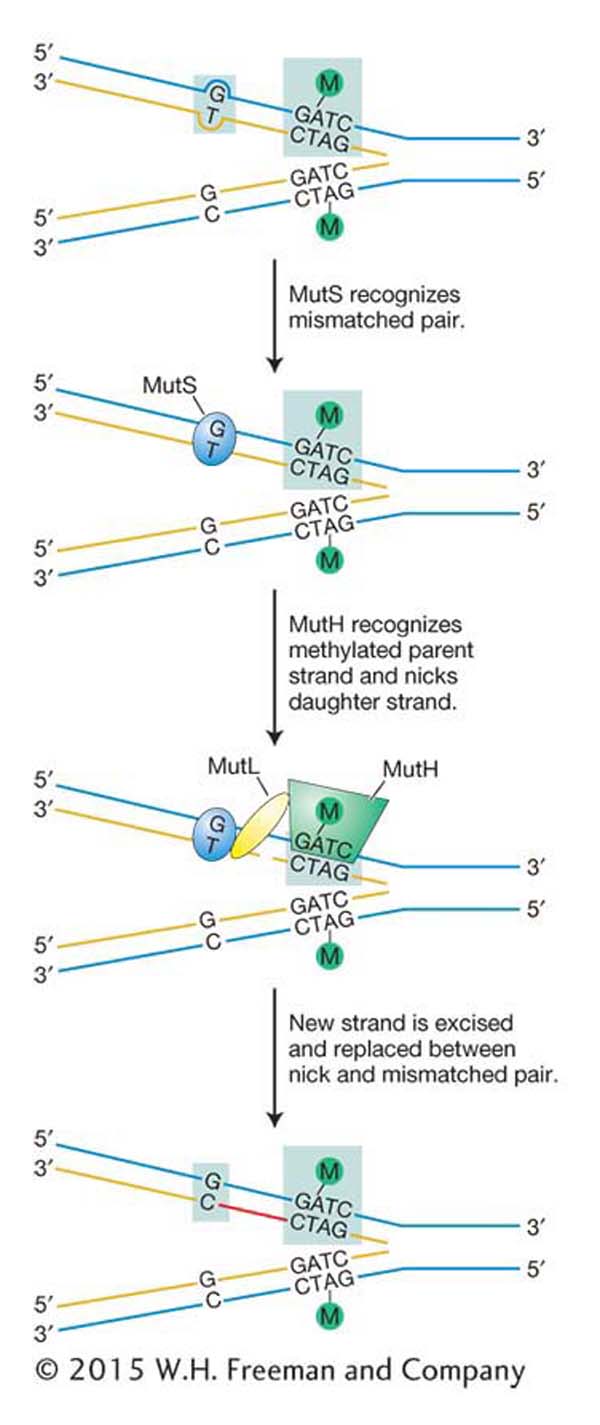
Figure 16-23: Mismatch repair corrects replicative errors
Figure 16-23: Model for mismatch repair in E. coli. DNA is methylated (Me) at the A residue in the sequence GATC. DNA replication yields a hemimethylated duplex that exists until methylase can modify the newly synthesized strand. The mismatch-repair system makes any necessary corrections based on the sequence found on the methylated strand (original template). MutS, MutH, and MutL are proteins.
Most of what is known about mismatch repair comes from decades of genetic and biochemical analysis with the use of the model bacterium E. coli (see the E. coli Model Organism box). Especially noteworthy was the reconstitution of the mismatch-repair system in the test tube in the laboratory of Paul Modrich. Conservation of many of the mismatch-repair proteins from bacteria to yeast to human indicates that this pathway is both ancient and important in all living organisms. Recently, the human mismatch-repair system also was reconstituted in the test tube in the Modrich laboratory. The ability to study the details of the reaction will spur future studies of the human pathway. However, for now we will focus on the very well characterized E. coli system (Figure 16-23).
The first step in mismatch repair is the recognition of the damage in newly replicated DNA by the MutS protein. The binding of this protein to distortions in the DNA double helix caused by mismatched bases initiates the mismatch-repair pathway by attracting three other proteins to the site of the lesion [MutL, MutH, and UvrD (not shown)]. The key protein is MutH, which performs the crucial function of cutting the strand containing the incorrect base. Without this ability to discriminate between the correct and the incorrect bases, the mismatch-repair system could not determine which base to excise to prevent a mutation from arising. If, for example, a G–T mismatch occurs as a replication error, how can the system determine whether G or T is incorrect? Both are normal bases in DNA. But replication errors produce mismatches on the newly synthesized strand, and so the mismatch-repair system replaces the base on that strand.
How does mismatch repair distinguish the newly synthesized strand from the old one? Recall from Chapter 12 that cytosine bases are often methylated in eukaryotes and that this so-called epigenetic mark is propagated from parent to daughter strand soon after replication. E. coli DNA also is methylated, but the methyl groups relevant to mismatch repair are added to adenine bases. To distinguish the old template strand from the newly synthesized strand, the bacterial repair system takes advantage of a delay in the methylation of the following sequence:
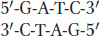
The methylating enzyme is adenine methylase, which creates 6-methyladenine on each strand. However, adenine methylase requires several minutes to recognize and modify the newly synthesized GATC stretches. In that interval, the MutH protein nicks the methylation site on the strand containing the A that has not yet been methylated. This site can be several hundred base pairs away from the mismatched base. After the site has been nicked, the UrvD protein binds at the nick and uses its helicase activity to unwind the DNA. A protective single-strand-binding protein coats the unwound parental strand while the part of the new strand between the mismatch and the nick is excised.
Many of the proteins in E. coli mismatch repair are conserved in human mismatch repair. Nonetheless, how eukaryotes recognize and repair only the newly replicated strand is still unknown. The problem is particularly perplexing in organisms that lack most or all DNA methylation such as yeast, Drosophila, and C. elegans. A popular model proposes that discrimination is based on the recognition of free 3′ ends that characterize the newly synthesized leading and lagging strands.
An important target of the human mismatch system is short repeat sequences that can be expanded or deleted in replication by the slipped-mispairing mechanism described previously (see Figure 16-8). Mutations in some of the components of this pathway have been shown to be responsible for several human diseases, especially cancers. There are thousands of short repeats (microsatellites) located throughout the human genome (see Chapter 4). Although most are located in noncoding regions (given that most of the genome is noncoding), a few are located in genes that are critical for normal growth and development.
Therefore, defects in the human mismatch-repair pathway would be predicted to have very serious disease consequences. This prediction has turned out to be true, a case in point being a syndrome called hereditary nonpolyposis colorectal cancer (HNPCC), which, despite its name, is not a cancer itself but increases cancer risk. One of the most common inherited predispositions to cancer, the disease affects as many as 1 in 200 people in the Western world. Studies have shown that HNPCC results from a loss of the mismatch-repair system due in large part to inherited mutations in genes that encode the human counterparts (and homologs) of the bacterial MutS and MutL proteins (see Figure 16-23). The inheritance of HNPCC is autosomal dominant. Cells with one functional copy of the mismatch-repair genes have normal mismatch-repair activity, but tumor cell lines arise from cells that have lost the one functional copy and are thus mismatch deficient. These cells display high mutation rates owing in part to an inability to correct the formation of indels in replication.
KEY CONCEPT
The mismatch-repair system corrects errors in replication that are not corrected by the proofreading function of the replicative DNA polymerase. Repair is restricted to the newly synthesized strand, which is recognized by the repair machinery in prokaryotes because it lacks a methylation marker.
Error-prone repair: translesion DNA synthesis
Thus far, all of the repair mechanisms that we have encountered are error free, inasmuch as they either reverse the damage directly or use base complementarity to insert the correct base. Yet, there are repair pathways that are themselves a significant source of mutation. These mechanisms appear to have evolved to prevent the occurrence of potentially more serious outcomes such as cell death or cancer. As already mentioned, a stalled replication fork can initiate a cell-death pathway. In both prokaryotes and eukaryotes, such replication blocks can be bypassed by the insertion of nonspecific bases. In E. coli, this process requires the activation of the SOS system. The name SOS comes from the idea that this system is induced as an emergency response to prevent cell death in the presence of significant DNA damage. As such, SOS induction is a mechanism of last resort, a form of damage tolerance that allows the cell to trade death for a certain level of mutagenesis.
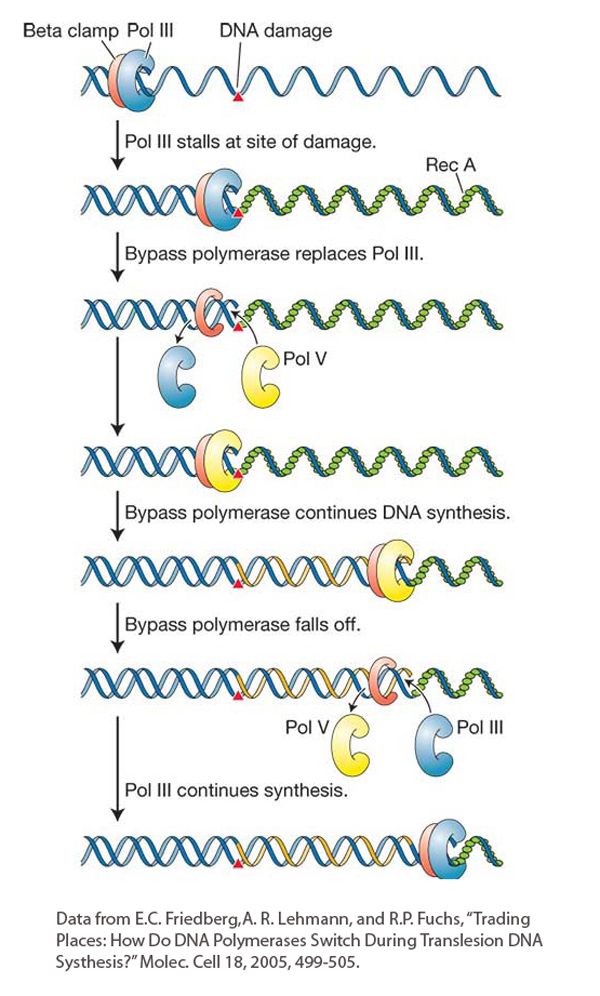
Figure 16-24: Translesion synthesis bypasses lesions at stalled replication forks
Figure 16-24: A model for translesion synthesis in E. coli. In the course of replication, DNA polymerase III is temporarily replaced by a bypass polymerase (pol V) that can continue replicating past a lesion. Bypass polymerases are error prone. The bacterial β clamp (red protein) is equivalent to the eukaryotic PCNA.
[Data from E. C. Friedberg, A. R. Lehmann, and R. P. Fuchs, “Trading Places: How Do DNA Polymerases Switch During Translesion DNA Synthesis?” Molec. Cell 18, 2005, 499–505.]
It has taken more than 30 years to figure out how the SOS system generates mutations while allowing DNA polymerase to bypass lesions at stalled replication forks. We are already familiar with DNA damage induced by UV light (see Figure 16-15). An unusual class of E. coli mutants that survived UV exposure without sustaining additional mutations was isolated in the 1970s. The fact that such mutants even existed suggested that some E. coli genes function to generate mutations when exposed to UV light. UV-induced mutation will not occur if the DinB, UmuC, or UmuD′ genes are mutated.
Figure 16-24 shows the steps in the SOS mechanism. In the first step, UV light induces the synthesis of a protein called RecA. We will see more of the RecA protein later in the chapter because it is a key player in key mechanisms of DNA repair and recombination. When the replicative polymerase (DNA polymerase III) stalls at a site of DNA damage, the DNA ahead of the polymerase continues to be unwound, exposing regions of single-stranded DNA that become bound by single-strand-binding proteins. Next, RecA proteins join the single-strand-binding proteins and form a protein–DNA filament. The RecA filament is the biologically active form of this protein. In this situation, RecA acts as a signal that leads to the induction of several genes that are now known to encode members of a newly discovered family of DNA polymerases that can bypass the replication block and are distinct from replicative polymerases. DNA polymerases that can bypass replication stalls have also been found in diverse taxa of eukaryotes ranging from yeast to human. These eukaryotic polymerases contribute to a damage-tolerance mechanism called translesion DNA synthesis that resembles the SOS bypass system in E. coli.
These translesion, or bypass polymerases, as they have come to be known, differ from the main replicative polymerases in several ways. First, they can tolerate unusually large adducts on the bases. Whereas the replicative polymerase stalls if a base does not fit into an active site, the bypass polymerases have much larger pockets that can accommodate damaged bases. Second, in some situations, the bypass polymerases have a much higher error rate, in part because they lack the 3′-to-5′ proofreading activity of the main replicative polymerases. Third, they can only add a few nucleotides before falling off. This feature is attractive because the main function of an error-prone polymerase is to unblock the replication fork, not to synthesize long stretches of DNA that could contain many mismatches.
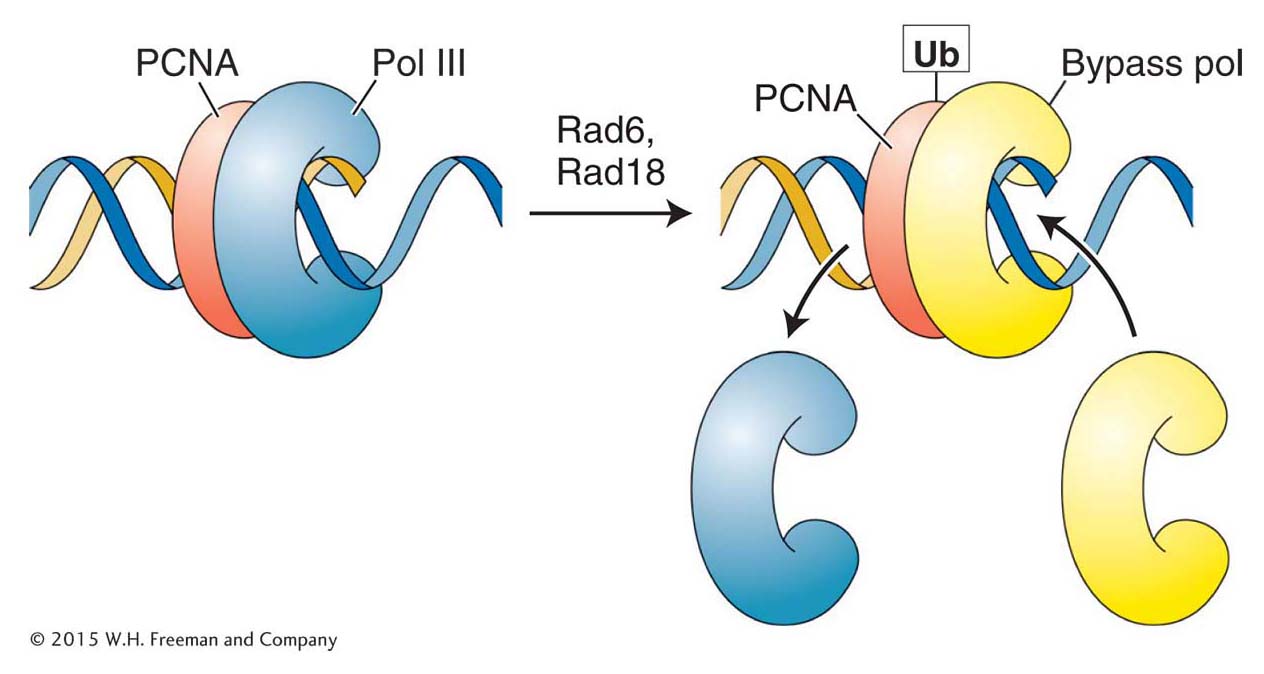
Figure 16-25: Polymerase switching requires addition of a ubiquitin monomer
Figure 16-25: The addition of a single ubiquitin (Ub) monomer to the sliding clamp (PCNA) allows the bypass polymerase to bind to PCNA and begin replicating.
Several bypass polymerases that appear to be always present in eukaryotic cells are now known. Because they are always present, their access to DNA must be regulated so that they are used only when needed. The cell has evolved a neat solution to this problem. Recall that an integral part of the replisome is the PCNA (proliferating cell nuclear antigen) protein that functions as a sliding clamp to orchestrate the myriad events at the replication fork (see Figure 7-20). One critical protein present at a stalled replication fork is Rad6, which, curiously, is an enzyme that adds ubiquitin to proteins (Figure 16-25). As described in Chapter 9, the addition of chains of many ubiquitin monomers serves to target a protein for degradation (see Figure 9-23). In contrast, the binding of a single ubiquitin monomer to PCNA changes its conformation so that it can now bind the bypass polymerase and orchestrate translesion synthesis. Enzymatic removal of the ubiquitin tag on PCNA leads to the dissociation of the bypass polymerase and the eventual restoration of normal replication. Any base mismatch due to translesion synthesis still has a chance of detection and correction by the mismatch-repair pathway.
The regulation of PCNA function by the addition and removal of ubiquitin monomers illustrates the importance of post-translational modifications in eukaryotes. If base damage in the template strand is not corrected quickly, the stalled replication fork will signal the activation of the cell-death pathway. A eukaryotic cell cannot wait for the de novo synthesis of bypass polymerases following transcription and translation as occurs in the E. coli SOS system. Instead, eukaryotic bypass polymerases are constitutively transcribed and are always present; their access to the replication fork is controlled by rapid and reversible post-translational modifications.
KEY CONCEPT
In translesion synthesis, bypass polymerases are recruited to replication forks that have stalled because of damage in the template strand. Bypass polymerases may introduce errors in the course of synthesis that may persist to mutation or that can be corrected by other mechanisms such as mismatch repair.
Repair of double-strand breaks
As we have seen, many correction systems exploit DNA complementarity to make error-free repairs. Such error-free repair is characterized by two stages: (1) removal of the damaged bases, perhaps along with nearby DNA, from one strand of the double helix and (2) use of the other strand as a template for the DNA synthesis needed to fill the single-strand gap. However, what would happen if both strands of the double helix were damaged in such a way that complementarity could not be exploited? For example, exposure to X-rays often causes both strands of the double helix to break at sites that are close together. This type of mutation is called a double-strand break. If left unrepaired, double-strand breaks can cause a variety of chromosomal aberrations resulting in cell death or a precancerous state.
Interestingly, the generation of double-strand breaks is an integral feature of some normal cellular processes that require DNA rearrangements. One example is meiotic recombination. As will be seen in the remainder of this chapter, the cell uses many of the same proteins and pathways to repair double-strand breaks and to carry out meiotic recombination. For this reason, we begin by focusing on the molecular mechanisms that repair double-strand breaks before turning our attention to the mechanism of meiotic recombination.
Double-strand breaks can arise spontaneously (for example, in response to reactive oxygen species produced as a by-product of cellular metabolism) or they can be induced by ionizing radiation. Several mechanisms are known to repair double-strand breaks, and new mechanisms are still being discovered. Two distinct mechanisms are described in the following section: nonhomologous end joining and homologous recombination.
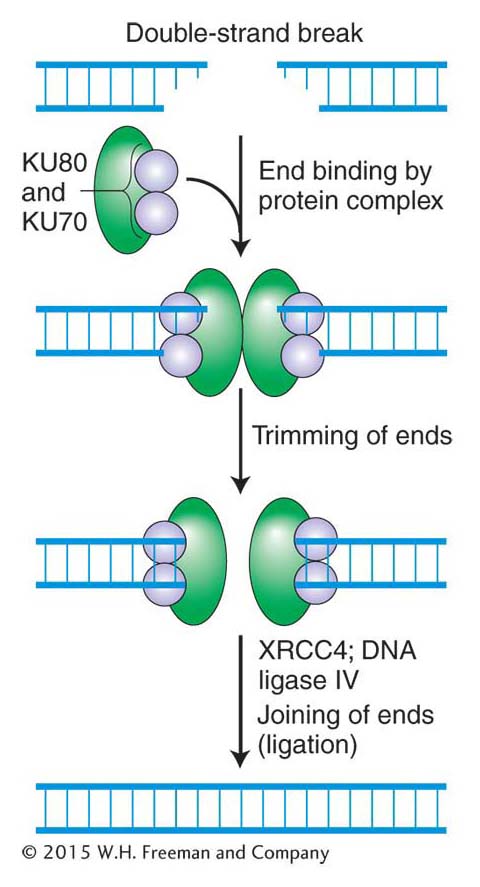
Figure 16-26: Error-prone nonhomologous end joining repairs double-strand breaks
Figure 16-26: Mechanism of nonhomologous end joining (NHEJ). This mechanism is error prone. See text for details.
Nonhomologous end joining Many of the previously described repair mechanisms are called on in the S phase of the cell cycle, when the DNA is replicating in preparation for mitosis or meiosis. However, unlike the cells of most prokaryotes and lower eukaryotes, the cells of higher eukaryotes are usually not replicating their DNA, because they are either in a resting phase of the cell cycle or have ceased dividing entirely. What happens when double-strand breaks occur in cells where undamaged strands or sister chromatids are not present? The answer is that these ends must be repaired, either perfectly or imperfectly, because broken ends can initiate potentially harmful chromosomal rearrangements that could lead to a cancerous state (see Chapter 17).
One way that higher eukaryotes put double-stranded broken ends back together is by a rather inelegant but important mechanism called nonhomologous end joining (NHEJ), which is shown in Figure 16-26. Like that of other repair mechanisms, the first step in the NHEJ pathway is recognition of the damage. The NHEJ pathway is initiated when two very abundant proteins, KU70 and KU80, bind to the broken ends, forming a heterodimer that serves two functions. First, it prevents further damage to the ends, and, second, it recruits other proteins (green, in Figure 16-26) that trim the strand ends to generate the 5′-P and 3′-OH ends that are required for ligation. DNA ligase IV then joins the two ends.
How do scientists know when all of the components of a biological pathway have been identified? As it turns out, this problem is difficult. The recent identification of a new component of the NHEJ pathway provides an example.
For several reasons, all of the components of the NHEJ pathway were thought to have been identified. However, geneticists analyzing a cell line (called 2BN) derived from a child with a rare inherited disorder were in for a surprise. Although they were able to demonstrate that cell line 2BN was defective for double-strand-break repair, they were not able to restore the repair system and produce the wild-type phenotype by genetic complementation with any of the genes encoding NHEJ proteins. That is, when they introduced wild-type genes encoding known NHEJ proteins (for example, KU70, KU80, ligase IV) into the 2BN line, the cell line was still defective in the repair of double-strand breaks. This negative result indicated that cell line 2BN carried a mutation in an unknown NHEJ protein.
In the era of genomics, the identification of proteins linked to diseases is becoming more common because of the wide availability of cell lines from persons with disease phenotypes. When we humans have a health problem, we go to a doctor and tell him or her about our symptoms, including information about relatives with similar problems. Such information is of increasing importance in the genomics era with its ever-expanding genetic toolbox that can often be used to identify mutant genes associated with inherited disorders (see Chapters 10 and 14).
WHAT GENETICISTS ARE DOING TODAY
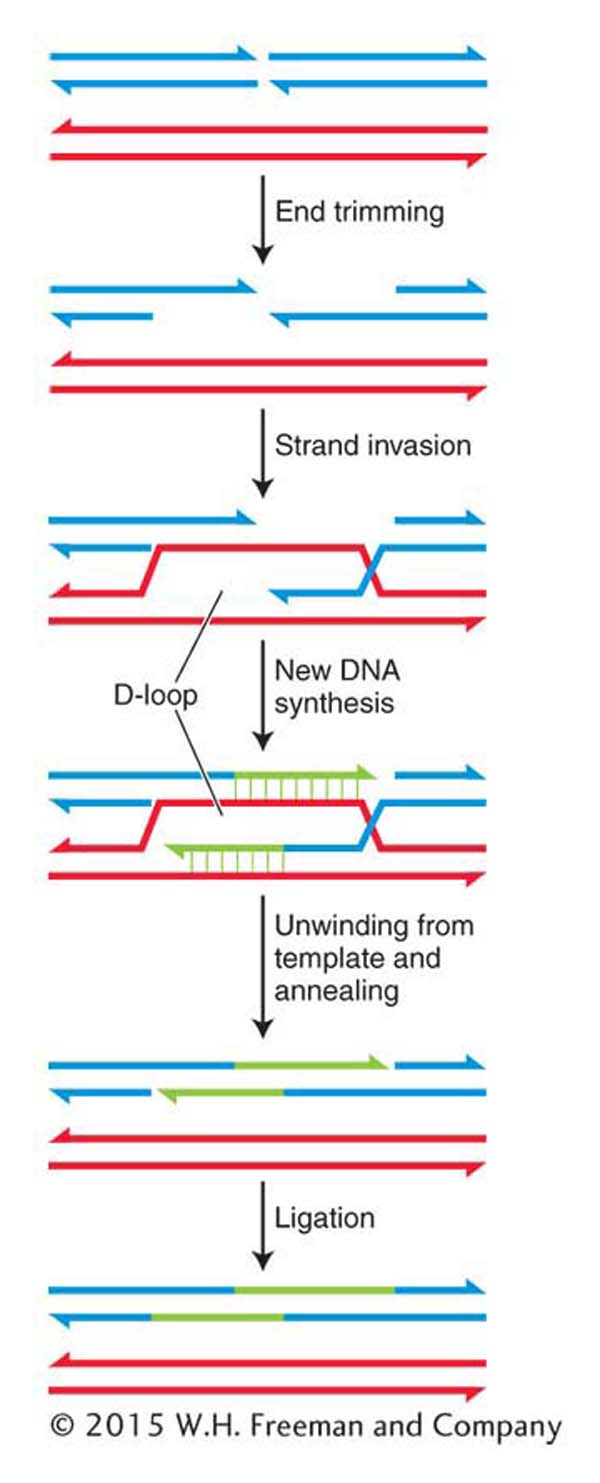
Figure 16-27: Error-free repair of double-strand breaks by SDSA
Figure 16-27: The error-free mechanism of synthesis-dependent strand annealing (SDSA) repairs double-strand breaks in dividing cells.
What can geneticists do in the laboratory to find a protein, such as the unknown NHEJ protein, that has not yet been identified? Two laboratories using very different approaches succeeded in identifying the new NHEJ component; one approach will be described here because it has been successfully employed to discover several other proteins. As noted in preceding chapters, many cellular proteins perform their jobs by interacting with other proteins. Chapter 14, for example, described the yeast two-hybrid test used to identify proteins that interact with a protein of interest. In the case under consideration now, the protein of interest was the NHEJ component XRCC4 (see Figure 16-26), and the two-hybrid test identified a 33-kD interacting protein that was encoded by an uncharacterized human open reading frame. That two proteins interact in the yeast two-hybrid test does not necessarily mean that these proteins interact in human cells. To establish a connection between the 33-kD protein and the NHEJ pathway, the geneticists used another valuable technique from their toolbox, RNAi (see Chapter 8). In this case, they demonstrated that normal cells expressing antisense RNA from the ORF that encodes the 33-kD protein, which would prevent translation of this gene into protein, were now defective in the execution of the NHEJ pathway.
This story came full circle when the 2BN cells defective in double-strand repair were shown to lack the 33-kD protein. Expression of this protein corrected the cellular defects.
KEY CONCEPT
NHEJ is an error-prone pathway that repairs double-strand breaks in higher eukaryotes by ligating the free ends back together. The identification of genes responsible for inherited disorders is an important way used by geneticists to isolate formerly unknown components of repair and other biological pathways.Homologous recombination If a double-strand break occurs after replication of a chromosomal region in a dividing cell, the damage can be corrected by an error-free mechanism called synthesis-dependent strand annealing (SDSA). This mechanism is depicted in Figure 16-27. It uses the sister chromatids available in mitosis as the templates to ensure correct repair.
The first steps in SDSA are the binding of the broken ends by specialized proteins and enzymes, the trimming of the 5′ ends by an endonuclease to expose single-stranded regions, and the coating of these regions with proteins that include the RecA homolog, Rad51. Recall that in the SOS response, RecA monomers associate with regions of single-stranded DNA to form nucleoprotein filaments. Similarly, Rad51 forms long filaments as it associates with the exposed single-stranded region. The Rad51–DNA filament then takes part in a remarkable search of the undamaged sister chromatid for the complementary sequence that will be used as a template for DNA synthesis. This process is called strand invasion. The 3′ end of the invading strand displaces one of the undamaged sister chromatids, which forms a D-loop (for displacement), and primes DNA synthesis from its free 3′ end. New DNA synthesis continues from both 3′ ends until both strands unwind from their templates and anneal. Ligation seals the nicks, leaving a repaired patch of DNA that has one very distinctive feature: it has been replicated by a conservative process. That is, both strands are newly synthesized, which stands in marked contrast to the semiconservative replication of most DNA (see Chapter 7).
KEY CONCEPT
Synthesis-dependent strand annealing is an error-free mechanism that repairs double-strand breaks in dividing cells in which a sister chromatid is available to serve as template for repair synthesis.
The involvement of DSB repair in meiotic recombination
Our consideration of the repair of double-strand breaks in dividing cells leads naturally to the topic of crossing over at meiosis because a double-strand break initiates the crossover event. Although the breaks are a normal and essential part of meiosis, they are, if not processed correctly and efficiently, as dangerous as the accidental breaks discussed so far.
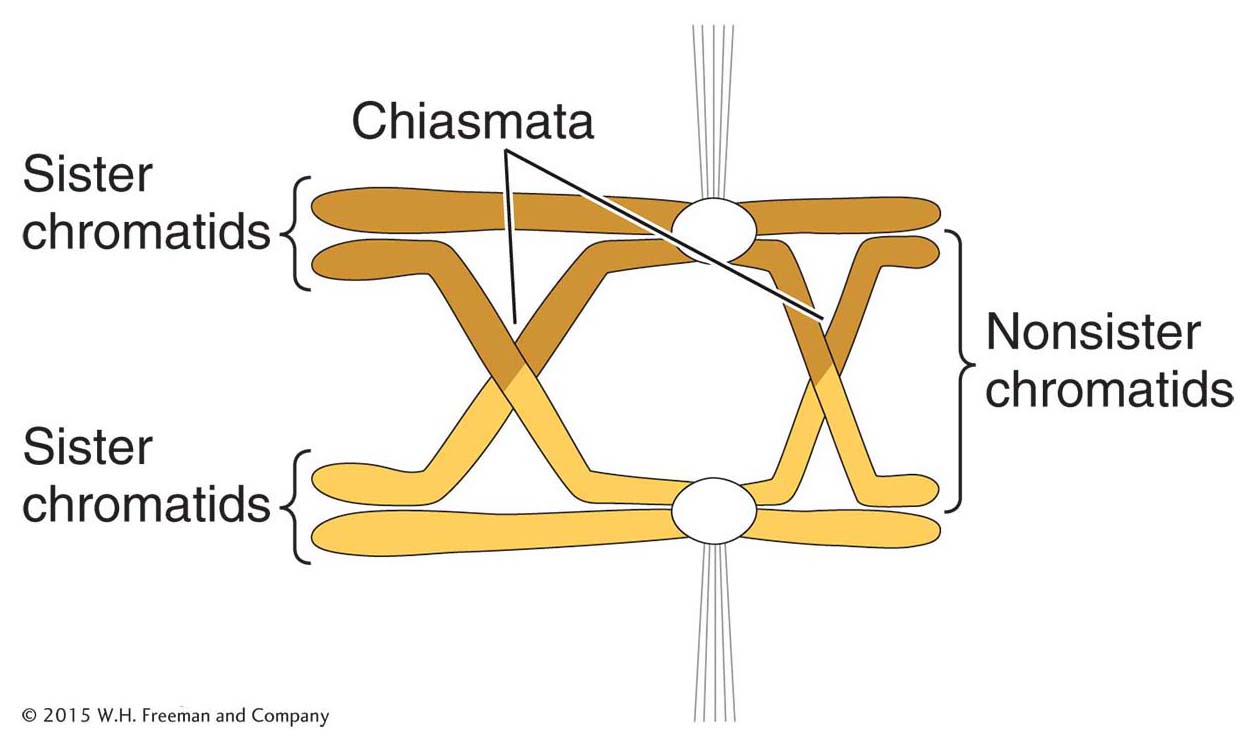
Figure 16-28: Crossing over during meiosis
Figure 16-28: Exchange of chromosome arms between nonsister chromatids during meiosis yields a chiasma, the location of crossovers. Circles represent centromeres that are attached to the spindle fibers.
Crossing over is a remarkably precise process that takes place between two homologous chromosomes (Figure 16-28). That process was described in Section 4.8. Recall that recombination takes place after the replication fork has passed through a chromosomal region, forming two chromatids from each homologous chromosome. One chromatid from one homologous chromosome will recombine with a nonsister chromatid from the other homologous chromosome. For meiotic segregation to work correctly, every pair of homologs must have at least one crossover. Recombination is initiated when an enzyme called Spo11 makes DNA double-strand cuts in one of the chromatids that will recombine (Figure 16-29). Although first discovered in yeast, the Spo11 protein is widely conserved in eukaryotes, indicating that this mechanism to initiate recombination is widely employed.
After making its cuts, the Spo11 enzyme remains attached to the now free 5′ ends, where it appears to serve two purposes. First, it protects the ends from further damage, including spurious recombination with other free ends. Second, it may attract other proteins that are needed for the next step in recombination. That step is actually very similar to what happens in the repair of double-strand breaks in dividing cells. The 5′ ends are trimmed back (resected), and a protein complex binds to the single-stranded 3′ ends (see Figure 16-29). That complex includes the Rad51 protein, which, as already mentioned, is a homolog to the RecA protein that takes part in that remarkable search for complementarity in the sister chromatid.
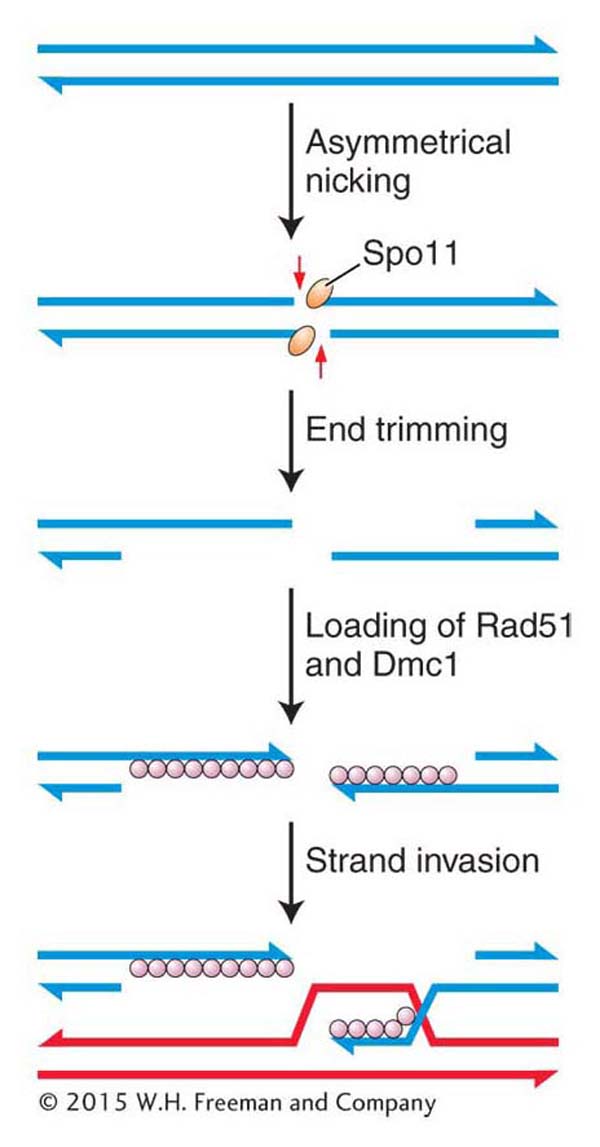
Figure 16-29: Double-strand breaks initiate meiotic recombination
Figure 16-29: Meiotic recombination is initiated when the enzyme Spoii makes staggered nicks in a pair of DNA strands in a chromatid.
At this time, meiotic recombination takes a dramatically different path from double-strand-break repair. In meiosis, Rad51 associates with another protein, Dmc1, which is present only during meiosis (see Figure 16-29). (It should be noted that the model organisms Drosophila and C. elegans do not have Dmc1 homologs.) Somehow, by an incompletely understood mechanism, the filament containing Rad51–Dmc1 conducts a search for a complementary sequence. However, in contrast with double-strand-break repair, the filament searches a nonsister chromatid from the homologous chromosome, not the sister chromatid. The search culminates in strand invasion and D-loop formation, just as in double-strand-break repair. These events are necessary for chiasma formation in meiosis I. That is, the homologs become connected as a result of recombination.
KEY CONCEPT
Meiotic recombination is initiated by the Spoil enzyme, which introduces double-strand cuts into chromosomes after they have replicated but before homologs separate.