How cancer cells differ from normal cells
A malignant tumor, or cancer, is an aggregate of cells, all descended from an initial aberrant founder cell. In other words, the malignant cells are all members of a single clone, even in advanced cancers having multiple tumors at many sites in the body. Cancer cells typically differ from their normal neighbors by a host of phenotypic characters, such as rapid division rate, ability to invade new cellular territories, high metabolic rate, and abnormal shape. For example, when cells from normal epithelial cell sheets are placed in cell culture, they can grow only when anchored to the culture dish itself. In addition, normal epithelial cells in culture divide only until they form a single continuous layer (Figure 16-30a). At that point, they somehow recognize that they have formed a single epithelial sheet and stop dividing. In contrast, malignant cells derived from epithelial tissue continue to proliferate, piling up on one another (Figure 16-30b).
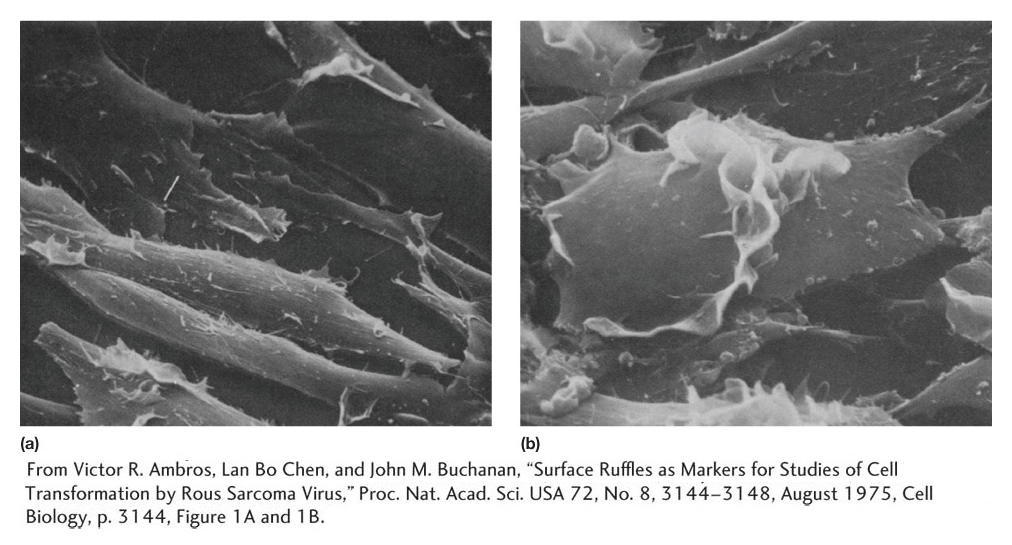
Figure 16-30: Normal cells and cells transformed by an oncogene
Figure 16-30: Scanning electron micrographs of (a) normal cells and (b) cells transformed by Rous sarcoma virus, which infects cells with the src oncogene. (a) A normal cell line called 3T3. Note the organized monolayer structure of the cells. (b) A transformed derivative of 3T3. Note how the cells are rounder and piled up on one another.
[From Victor R. Ambros, Lan Bo Chen, and John M. Buchanan, “Surface Ruffles as Markers for Studies of Cell Transformation by Rous Sarcoma Virus,” Proc. Nat. Acad. Sci. USA 72, No. 8, 3144-3148, August 1975, Cell Biology, p. 3144, Figure 1A and 1B.]
Clearly, the factors regulating normal cellular physiology have been altered. What, then, is the underlying cause of cancer? Many different cell types can be converted into a malignant state. Is there a common theme? Or does each arise in a quite different way? We can think about cancer in a general way as being due to the accumulation of multiple mutations in a single cell that cause it to proliferate out of control. Some of those mutations may be transmitted from the parents through the germ line. But most arise de novo in the somatic-cell lineage of a particular cell.
Mutations in cancer cells
Several lines of evidence point to a genetic origin for the transformation of cells from the benign into the cancerous state. First, as already discussed in this chapter, many mutagenic agents such as chemicals and radiation cause cancer, suggesting that they produce cancer by introducing mutations into genes. Second, and most importantly, mutations that are frequently associated with particular kinds of cancers have been identified.
Two general kinds are associated with tumors: oncogene mutations and mutations in tumor-suppressor genes. Oncogene mutations act in the cancer cell as gain-of-function dominant mutations (see Chapter 6 for a discussion of dominant mutations). That statement suggests two key characteristics of oncogene mutations. First, the proteins encoded by oncogenes are usually activated in tumor cells, and, second, the mutation need be present in only one allele to contribute to tumor formation. The gene in its normal, unmutated form is called a proto-oncogene.
Mutations in tumor-suppressor genes that promote tumor formation are loss-of-function recessive mutations. That is, this type of mutation causes the encoded gene products to lose much or all of their activity (that is, the mutation is a null mutation). Moreover, for cancer to develop, the mutation must be present in both alleles of the gene.
KEY CONCEPT
Oncogenes encode mutated forms of normal cellular proteins that result in dominant mutations, usually owing to their inappropriate activation. In contrast, tumor-suppressor genes encode proteins whose loss of activity can contribute to a cancerous state. As such, they are recessive mutations.Classes of oncogenes Roughly a hundred different oncogenes have been identified. How do their normal counterparts, proto-oncogenes, function? Proto-oncogenes generally encode a class of proteins that are active only when the proper regulatory signals allow them to be activated. Many proto-oncogene products are elements in pathways that induce (positively control) the cell cycle. These products include growth-factor receptors, signal-transduction proteins, and transcriptional regulators. Other proto-oncogene products act to inhibit (negatively control) the apoptotic pathway that destroys damaged cells. In both types of oncogene mutation, the activity of the mutant protein has been uncoupled from its normal regulatory pathway, leading to its continuous unregulated expression. The continuously expressed protein product of an oncogene is called an oncoprotein. Several categories of oncogenes have been identified according to the different ways in which the regulatory functions have been uncoupled.
The ras oncogene can be used to illustrate what happens when a normal gene sustains a tumor-promoting mutation. As is often the case, the change from normal protein to oncoprotein entails structural modifications of the protein itself— in this case, caused by a simple point mutation. A single base-pair substitution that converts glycine into valine at amino acid number 12 of the Ras protein, for example, creates the oncoprotein found in human bladder cancer (Figure 16-31a). The normal Ras protein is a G-protein subunit that takes part in signal transduction. It normally functions by cycling between the active GTP-bound state and the inactive GDP-bound state. The missense mutation in the ras oncogene produces an oncoprotein that always binds GTP (Figure 16-31b), even in the absence of normal signals. As a consequence, the Ras oncoprotein continuously propagates a signal that promotes cell proliferation.
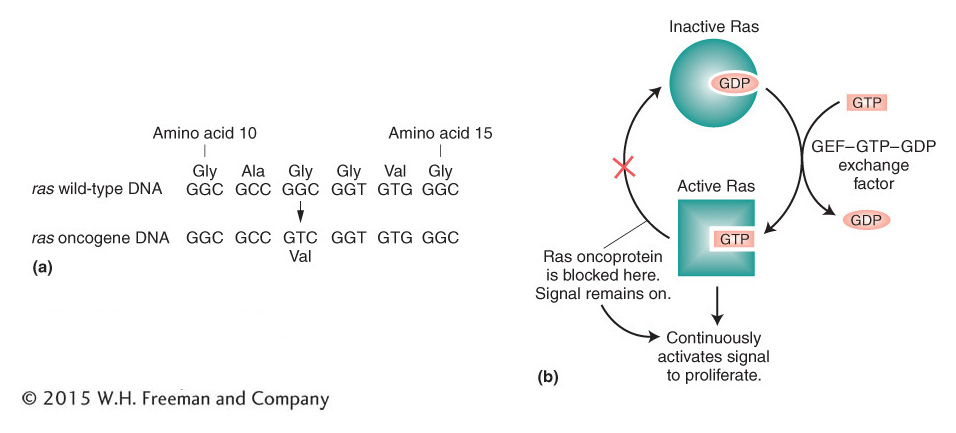
Figure 16-31: The ras oncogene is continuously active
Figure 16-31: Formation and effect of the Ras oncoprotein. (a) The ras oncogene differs from the wild type by a single base pair, producing a Ras oncoprotein that differs from wild type in one amino acid, at position 12 in the ras open reading frame. (b) The Ras oncoprotein cannot hydrolyze GTP to GDP. Because of this defect, the Ras oncoprotein remains in the active Ras-GTP complex and continuously activates the signal to proliferate.
Tumor-suppressor genes The normal functions of tumor-suppressor genes fall into categories complementary to those of proto-oncogenes (Table 16-1). Some tumor-suppressor genes encode negative regulators whose normal function is to inhibit the cell cycle. Others encode positive regulators that normally activate apoptosis, or cell death, of a damaged cell. Still others are indirect players in cancer, with a normal role in the repair of damaged DNA or in controlling cellular longevity. We will consider one example here.
Wild-type protein function |
Properties of tumor-promoting mutations |
Promotes cell-cycle progression |
Oncogene (gain of function) |
Inhibits cell-cycle progression |
Tumor-suppressor mutation (loss of function) |
|
|
Tumor-suppressor mutation (loss of function) |
|
|
Oncogene (gain of function |
|
|
Tumor-suppressor mutation (loss of function) |
Table 16-1: Functions of Wild-Type Proteins and Properties of Tumor-Promoting Mutations in the Corresponding Genes
Mutations in the p53 gene are associated with many types of tumors. In fact, estimates are that 50 percent of human tumors lack a functional p53 gene. The active p53 protein is a transcriptional regulator that is activated in response to DNA damage. Activated wild-type p53 serves double duty: it prevents the progression of the cell cycle until the DNA damage is repaired, and, under some circumstances, it induces apoptosis. If no functional p53 gene is present, the cell cycle progresses even if damaged DNA has not been repaired. The progression of the cell cycle into mitosis elevates the overall frequency of mutations, chromosomal rearrangements, and aneuploidy and thus increases the chances that other mutations that promote cell proliferation or block apoptosis will arise.
It is now clear that mutations able to elevate the mutation rate are important contributors to the progression of tumors in humans. These mutations are recessive mutations in tumor-suppressor genes that normally function in DNA-repair pathways. Mutations in these genes thus interfere with DNA repair. They promote tumor growth indirectly by elevating the mutation rate, which makes it much more likely that a series of oncogene and tumor-suppressor mutations will arise, corrupting the normal regulation of the cell cycle and programmed cell death. Large numbers of such tumor-suppressor-gene mutations have been identified, including some associated with heritable forms of cancer in specific tissues. Examples are the BRCA1 and BRCA2 mutations and breast cancer.
KEY CONCEPT
Mutagenic agents can cause some cancers because cancer is, in part, caused by mutant versions of normal genes that lead to uncontrolled growth.