18.1 Detecting Genetic Variation
The methods of population genetics can be used to analyze any variable or polymorphic locus in the DNA sequences of a population of organisms. Historically, geneticists lacked the molecular tools needed to observe differences in the DNA sequences among individuals directly, and so most population genetic analyses looked at differences in proteins or phenotypes. For example, differences in the protein encoded by the ABO glycosyltransferase gene controlling the ABO blood group in humans can be detected using antibody probes. From these protein differences, investigators can infer differences in the DNA sequence of this gene among individuals. Over the past three decades, new technologies, such as DNA sequencing, DNA microarrays, and PCR (see Chapters 10 and 14), have been developed that allow geneticists to observe differences in the DNA sequences directly. As a result, population genetic analyses are no longer confined to a small set of genes such as ABO but have expanded to include every nucleotide in the genome.
667
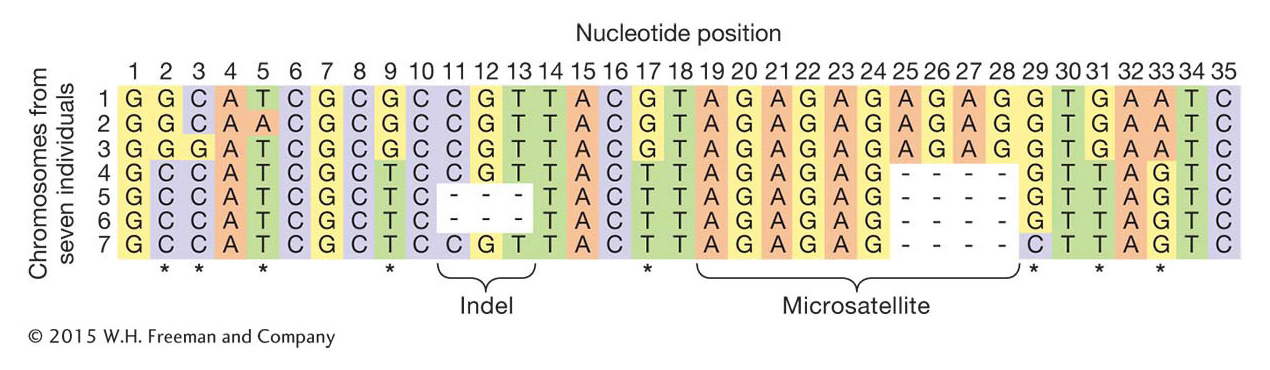
In population genetics, a locus is simply a location in the genome; it can be a single nucleotide site or a stretch of many nucleotides. The simplest form of variation one might observe among individuals at a locus is a difference in the nucleotide present at a single nucleotide site, whether adenine, cytosine, guanine, or thymine. These types of variants are called single nucleotide polymorphisms (SNPs), and they are the most widely studied variants in human population genetics (Figure 18-1; see also Chapter 4). Population genetics also makes extensive use of microsatellite loci (see Chapter 4). These loci have a short sequence motif, 2 to 6 base pairs long, that is repeated multiple times with different alleles having different numbers of repeats. For example, the 2-
Single nucleotide polymorphisms (SNPs)
SNPs are the most prevalent types of polymorphism in most genomes. Most SNPs have just two alleles—

SNPs occur within genes, including within exons, introns, and regulatory regions. SNPs within protein-
668
To study SNP variation in a population, we first need to determine which nucleotide sites in the genome are variable—
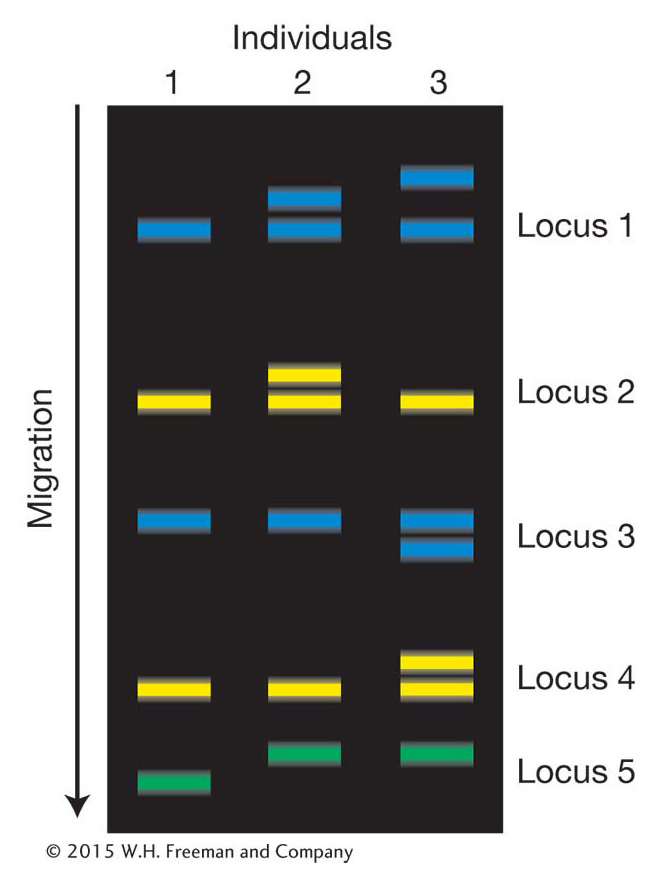
Once SNPs have been discovered, the genotype (allelic composition) of different individuals in the population at each SNP can be determined. DNA microarrays are a widely used technology for this purpose (Figure 18-2). The microarrays used for SNP assays can contain thousands of probes corresponding to known SNPs. Biotechnologists have developed several different methods to detect SNP variants using microarrays. In one method, DNA from an individual is labeled with fluorescent tags and hybridized to the microarray. Each spot (SNP) on the microarray will fluoresce red for one homozygous class, green for the other homozygote, and yellow for a heterozygote (see Figure 18-2). The entire procedure has been enhanced with robotics to allow rapid genotyping, or assignment of genotypes (for example, A/A versus A/C) on a large-
Microsatellites
Microsatellites are powerful loci for population genetic analysis for several reasons. First, unlike SNPs, which typically have only two alleles per locus and can never have more than four alleles, the number of alleles at a microsatellite is often very large (20 or more). Second, they have a high mutation rate, typically in the range of 10−3 to 10−4 mutations per locus per generation as compared to 10−8 to 10−9 mutations per site per generation for SNPs. The high mutation rate means that levels of variation are higher: more alleles per locus and a greater chance that any two individuals will have different genotypes. Third, microsatellites are very abundant in most genomes. Humans have over a million microsatellites.
Microsatellites are found throughout the genomes of most organisms and may be present in exons, introns, regulatory regions, and nonfunctional DNA sequences. Microsatellites with trinucleotide repeats are found in the coding sequences of some genes; these encode strings of a single amino acid. The Huntington disease gene (HD) (see Chapter 16) contains a repeat of CAG, which encodes a string of glutamines. Individuals carrying alleles with more than 30 glutamines are predisposed to develop the disease. In general, however, most microsatellites are located outside of coding sequences, and variation in the number of repeats is not associated with differences in phenotype.
Two main methods are used to discover microsatellite loci in the genome. If a complete genomic sequence is available for an organism, one can simply conduct a search to find them using a computer. For species without genome sequences (most non–
669
Once a microsatellite and its flanking sequences have been identified, DNA samples from a set of individuals in the population can be analyzed to determine the number of repeats that are present in each individual. To carry out the analysis, oligonucleotide primers are designed that match the flanking sequences for use in PCR. If the primers are labeled with a fluorescent tag, then the sizes of the PCR products can be determined on the same apparatus used to determine the sequence of DNA molecules (Figure 18-3). These sizes reveal the number of repeats in a microsatellite allele. For example, the PCR product of a microsatellite allele containing seven AG repeats will be 8 bp longer than an allele containing three AG repeats. Heterozygous individuals will possess products of two different sizes. Since PCR, the sizing of PCR products, and scoring of the alleles can all be automated, it is possible to determine the genotypes of large samples of individuals for large numbers of microsatellites relatively rapidly.
Haplotypes
For some questions in population genetics, it is important to consider the genotypes of linked loci as a group rather than individually. Geneticists use the term haplotype to refer to the combination of alleles at multiple loci on the same chromosomal homolog. Two homologous chromosomes that share the same allele at each of the loci under consideration have the same haplotype. If two chromosomes have different genotypes at even one of the loci in question, then they have different haplotypes. If the A locus with alleles A and a is linked to the B locus with alleles B and b, then there are four possible haplotypes for the chromosomal segment on which these two loci are located:

A more complex, but more realistic, example is shown in Figure 18-4. In Figure 18-4a, there are seven chromosome segments but only six haplotypes because chromosome segments 5 and 6 have the same haplotype (E).
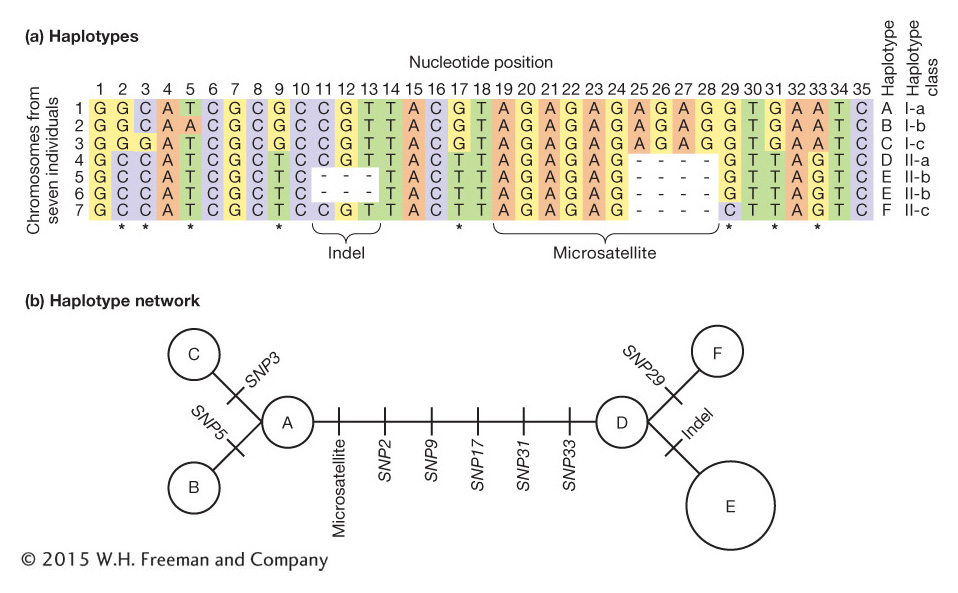
Haplotypes are most often used in population genetics for loci that are physically close. For example, the variable-
What insights can we gain from haplotype analysis? Population geneticists studying the human Y chromosome among Asian men discovered one highly prevalent haplotype, termed the “star-

670
Other sources and forms of variation
Beyond SNP and microsatellites, any variation in the DNA sequence of the chromosomes in a population is amenable to population genetic analysis. Variations that can be analyzed include inversions, translocations, deletions or duplications, and the presence or absence of a transposable element at a particular locus in the genome. Another common form of variation is insertion-
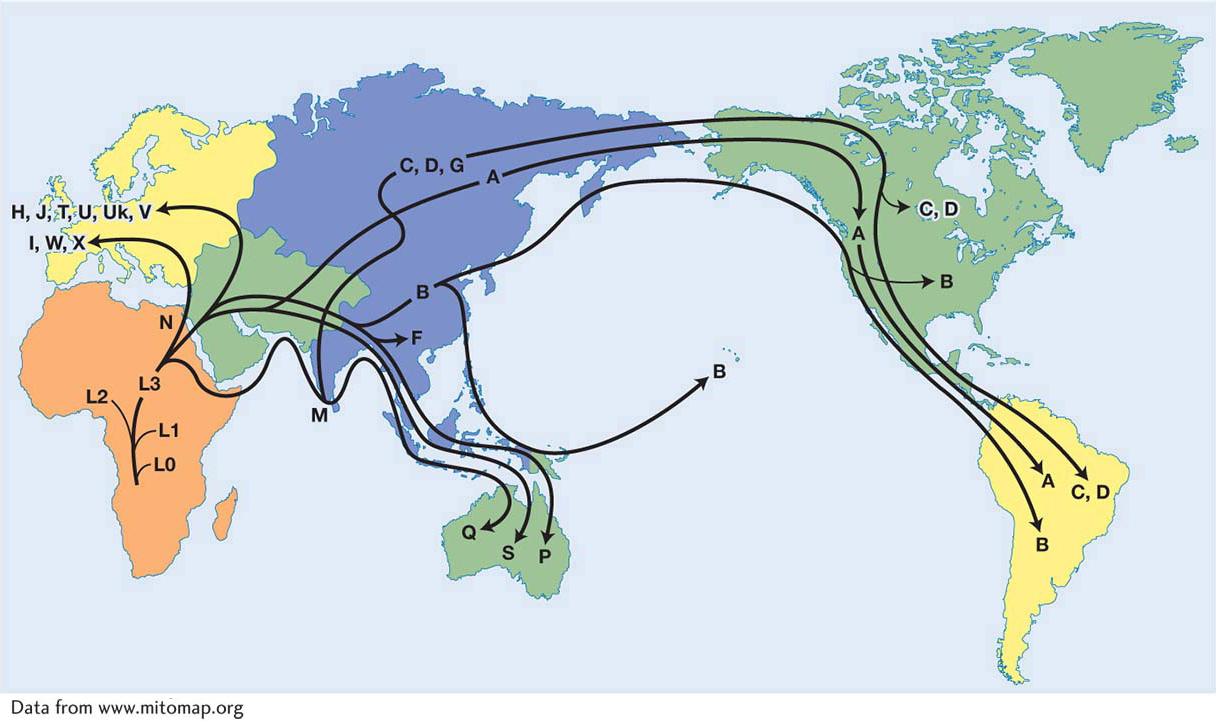
Thus far, our discussion of SNP and microsatellites has focused on the nuclear genome. However, interesting genetic variation can also be found in the mitochondrial (mtDNA) and chloroplast (cpDNA) genomes of eukaryotes. Both SNP and microsatellites are found in these organelle genomes. Since mtDNA and cpDNA are usually maternally inherited, their analysis can be used to follow the history of female lineages. In 1987, a prominent study of the human mitochondrial lineage traced the history of the human mtDNA haplotypes and determined that the mitochondrial genomes of all modern humans trace back to a single woman who lived in Africa about 150,000 years ago (Figure 18-6). She was dubbed the “mitochondrial Eve” in the popular press. This study of mtDNA was the first thorough genetic analysis to suggest that all modern humans came from Africa.
671
The HapMap Project
A major advance in human population genetics over the past decade was the creation of a genome-
672