19.5 Mapping QTL in Populations with Known Pedigrees
The genes that control variation in quantitative (or complex) traits are known as quantitative trait loci, or QTL for short. As we will see below, QTL are genes just like any others that you have learned about in this book. They may encode metabolic enzymes, cell-
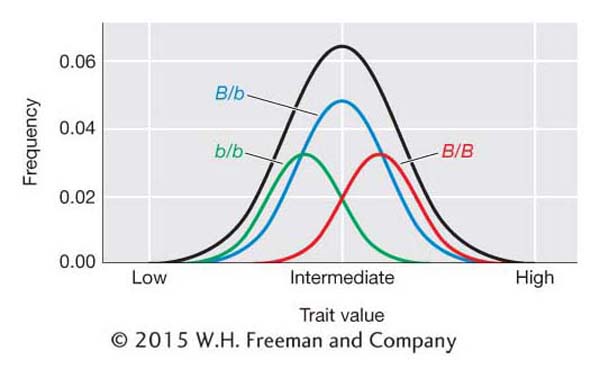
We can visualize the contributions of the alleles at a QTL to the trait value by looking at the frequency distributions associated with each genotype at a QTL as shown in Figure 19-11. The QTL locus is B and the genotypic classes are B/B, B/b, and b/b. The B/B individuals tend to have higher trait values, B/b intermediate values, and b/b small values. However, their distributions overlap, and we cannot determine genotype simply by looking at an individual’s phenotype as we can for genes that segregate in Mendelian ratios. In Figure 19-11, an individual with an intermediate trait value could be either B/B, B/b, or b/b.
Because of this property of QTL, we need special tools to determine their location in the genome and characterize their effects on trait variation. In this section, we will review a powerful form of analysis for accomplishing the first of these goals. This form of analysis is called QTL mapping. Over the past two decades, QTL mapping has revolutionized our understanding of the inheritance of quantitative traits. Pioneering work in QTL mapping was performed with crop plants such as tomato and corn. However, it has been broadly applied in model organisms such as mouse, Drosophila, and Arabidopsis. More recently, evolutionary biologists have employed QTL mapping to investigate the inheritance of quantitative traits in natural populations.
743
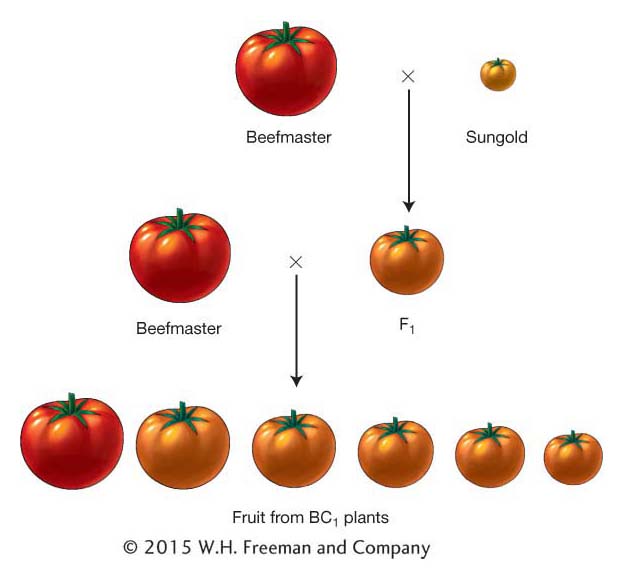
The fundamental idea behind QTL mapping is that one can identify the location of QTL in the genome using marker loci linked to a QTL. Here is how the method works. Suppose you make a cross between two inbred strains—
The basic method
There are a variety of experimental designs that can be used in QTL mapping experiments. We will begin by describing a simple design. Let’s say we have two inbred lines of tomato that differ in fruit weight—
744
From this process, we would assemble a data set for several hundred plants and 100 or more marker loci distributed around the genome. Table 19-6 shows part of such a data set for just 20 plants and 5 marker loci that are linked on a single chromosome. For each BC1 plant, we have the weight of its fruit and the genotypes at the marker loci. You’ll notice that trait values for the BC1 plants are intermediate between the two parents as expected but closer to the Beefmaster value because this is a BC1 population and Beefmaster was the backcross parent. Also, since this is a backcross population, the genotypes at each marker locus are either homozygous for the Beefmaster allele (B/B) or heterozygous (B/S). In Table 19-6, you can see the positions of crossovers between the marker loci that occurred during meiosis in the F1 parent of the BC1 generation. For example, plant BC1-001 has a recombinant chromosome with a crossover between marker loci M3 and M4.
|
Markers |
||||||
|---|---|---|---|---|---|---|
|
Plant |
Fruit wt. (g) |
M1 |
M2 |
M3 |
M4 |
M5 |
|
Beefmaster |
230 |
B/B |
B/B |
B/B |
B/B |
B/B |
|
Sungold |
10 |
S/S |
S/S |
S/S |
S/S |
S/S |
|
BC1-001 |
183 |
B/B |
B/B |
B/B |
B/S |
B/S |
|
BC1-002 |
176 |
B/S |
B/S |
B/B |
B/B |
B/B |
|
BC1-003 |
170 |
B/B |
B/S |
B/S |
B/S |
B/S |
|
BC1-004 |
185 |
B/B |
B/B |
B/B |
B/S |
B/S |
|
BC1-005 |
182 |
B/B |
B/B |
B/B |
B/B |
B/B |
|
BC1-006 |
170 |
B/S |
B/S |
B/S |
B/S |
B/B |
|
BC1-007 |
170 |
B/B |
B/S |
B/S |
B/S |
B/S |
|
BC1-008 |
174 |
B/S |
B/S |
B/S |
B/S |
B/S |
|
BC1-009 |
171 |
B/S |
B/S |
B/S |
B/B |
B/B |
|
BC1-010 |
180 |
B/S |
B/S |
B/B |
B/B |
B/B |
|
BC1-011 |
185 |
B/S |
B/B |
B/B |
B/S |
B/S |
|
BC1-012 |
169 |
B/S |
B/S |
B/S |
B/S |
B/S |
|
BC1-013 |
165 |
B/B |
B/B |
B/S |
B/S |
B/S |
|
BC1-014 |
181 |
B/S |
B/S |
B/B |
B/B |
B/S |
|
BC1-015 |
169 |
B/S |
B/S |
B/S |
B/B |
B/B |
|
BC1-016 |
182 |
B/B |
B/B |
B/B |
B/S |
B/S |
|
BC1-017 |
179 |
B/S |
B/S |
B/B |
B/B |
B/B |
|
BC1-018 |
182 |
B/S |
B/B |
B/B |
B/B |
B/B |
|
BC1-019 |
168 |
B/S |
B/S |
B/S |
B/B |
B/B |
|
BC1-020 |
173 |
B/B |
B/B |
B/B |
B/B |
B/B |
|
Mean of B/B |
- |
176.3 |
179.6 |
180.7 |
176.1 |
175.0 |
|
Mean of B/S |
- |
175.3 |
173.1 |
169.6 |
175.3 |
176.4 |
|
Overall mean |
175.7 |
|
|
|
|
|
The overall mean fruit weight for the BC1 population is 175.7. We can also calculate the mean for the two genotypic classes at each marker locus as shown in Table 19-6. For marker M1, the means for the B/B (176.3) and B/S (175.3) genotypic classes are very close to the overall mean (175.7). This is the expectation if there is no QTL affecting fruit weight near M1. For marker M3, the means for the B/B (180.7) and B/S (169.6) genotypic classes are quite different from the overall mean (175.7) and from each other. This is the expectation if there is a QTL affecting fruit weight near M3. Thus, we have evidence for a QTL affecting fruit weight near marker M3. Also notice that the B/B class has heavier fruit than the B/S class of M3. Plants that inherited the S allele from the small-
745
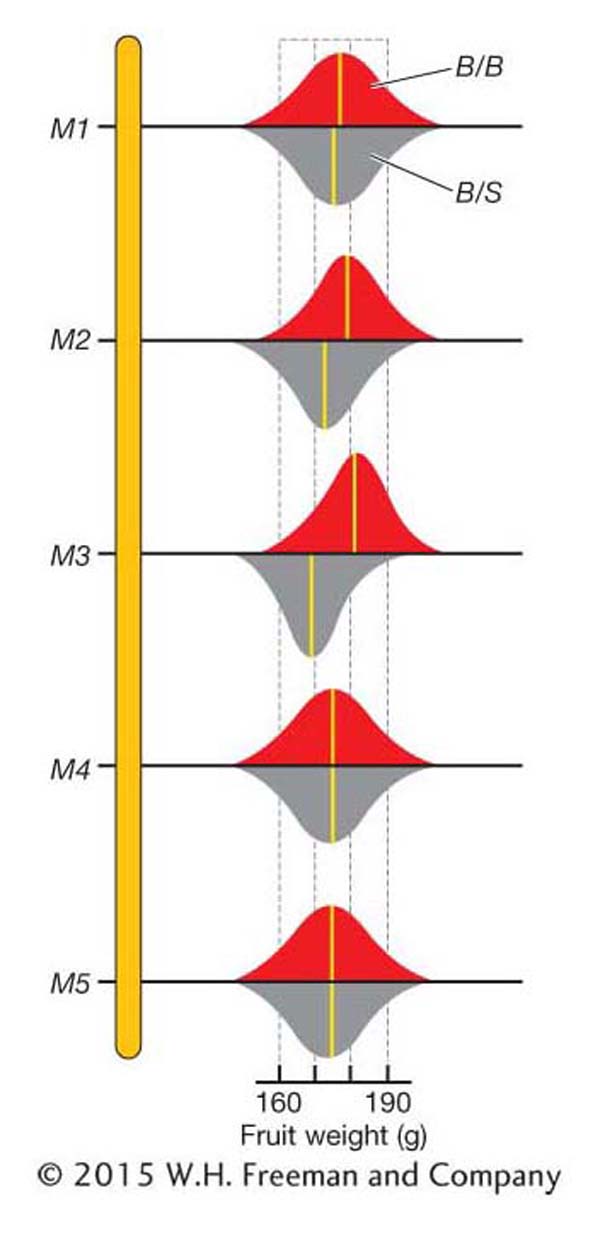
Figure 19-13 is a graphical representation of QTL-
As shown in Figure 19-13, the trait means for the B/B and B/S groups at some markers are nearly the same. At other markers, these means are rather different. How different do they need to be before we declare that a QTL is located near a marker? The statistical details for answering this question are beyond the scope of this text. However, let’s review the basic logic behind the statistics. The statistical analysis involves calculating the probability of observing the data (the specific fruit weights and marker-

The vertical line | means “given,” and the term Prob(data|QTL) reads “the probability of observing the data given that there is a QTL.” If the probability of the data when there is a QTL is 0.1 and the probability of the data when there is no QTL is 0.001, then the odds are 0.1/0.001 = 100. That is, the odds are 100 to 1 in favor of there being a QTL. Researchers report the log10 of the odds, or the Lod score. So, if the odds ratio is 100, then the log10 of 100, or Lod score, is 2.0.
If there is a QTL near the marker, then the data were drawn from two underlying distributions—
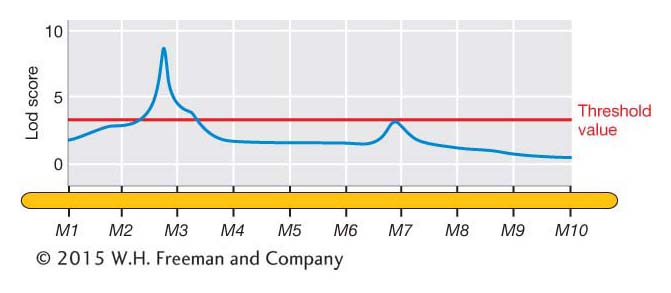
In addition to testing for QTL at the marker loci where the genotypes are known, Lod scores can be calculated for points between the markers. This can be done by using the genotypes of the flanking markers to infer the genotypes at points between the markers. For example, in Table 19-6, plant BC1-001 is B/B at markers M1 and M2, and so it has a high probability of being B/B at all points in between. Plant BC1-003 is B/B at marker M1 but B/S at M2, and so the plant might be either B/B or B/S at points in between. The odds equation incorporates this uncertainty when one calculates the Lod score at points between the markers.
The Lod scores can be plotted along the chromosome as shown by the blue line in Figure 19-14. Such plots typically show some peaks of various heights as well as stretches that are relatively flat. The peaks represent putative QTL, but how high does a peak need to be before we declare that it represents a QTL? As discussed in Chapters 4 and 18, we can set a statistical threshold for rejecting the “null hypothesis.” In this case, the null hypothesis is that “there is not a QTL at a specific position along the chromosome.” The greater the Lod score, then the lower the probability under the null hypothesis. There are different statistical procedures for setting a “threshold value” for the Lod score. Where the Lod score exceeds the threshold value, then we reject the null hypothesis in favor of the alternative hypothesis that a QTL is located at that position. In Figure 19-14, the Lod score exceeds the threshold value (red line) near marker locus M3. We conclude that a QTL is located near M3.
746
In addition to backcross populations, QTL mapping can be done with F2 populations and other breeding designs. An advantage of using an F2 population is that one gets estimates of the mean trait values for all three QTL genotypes: homozygous parent-
Here is an example. Suppose we studied an F2 population from a cross of Beef-
|
Fruit weights |
Effects |
|||||
|---|---|---|---|---|---|---|
|
B/B |
B/S |
S/S |
A |
D |
||
|
QTL 1 |
180 |
170 |
160 |
10 |
0 |
|
|
QTL 2 |
200 |
185 |
110 |
45 |
30 |
|
We can use these fruit weight values for the QTL to calculate the additive and dominance effects. QTL 1 is purely additive (D = 0), but QTL 2 has a large dominance effect. Also, notice that the additive effect of QTL 2 is more than 4 times that of QTL 1 (45 versus 10). Some QTL have large effects, and others have rather small effects.
What can be learned from QTL mapping? With the most powerful QTL-
Much has been learned about genetic architecture from QTL-
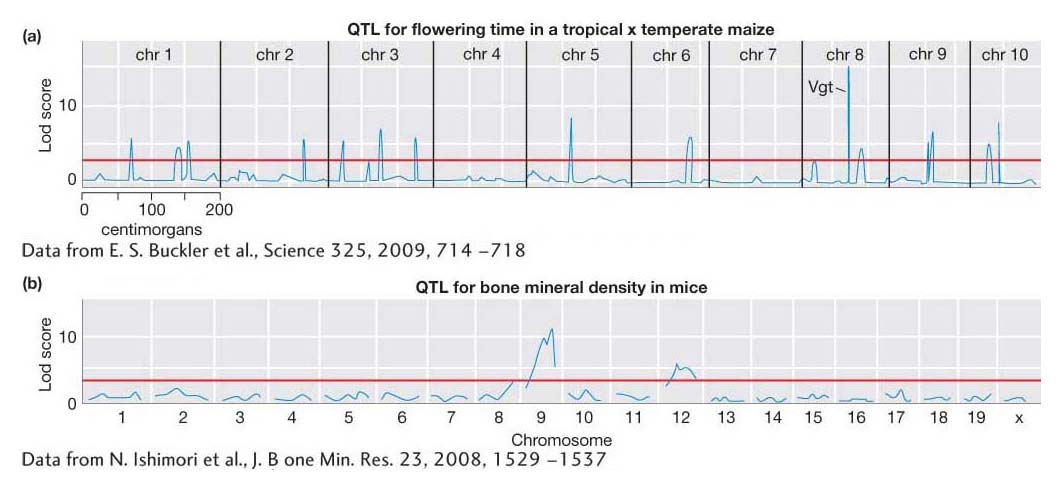
747
Second, mice have been used to map QTL for many disease-
From QTL to gene
QTL mapping does not typically reveal the identity of the gene(s) at the QTL. At its best, the resolution of QTL mapping is on the order of 1 to 10 cM, the size of a region that can contain 100 or more genes. To go from QTL to a single gene requires additional experiments to fine-
748
Using the tomato fruit weight example from above, the chromosome region for a set of such congenic lines is shown in Figure 19-16. The genes (flc, arf4,…) are shown at the top, and the location for each crossover is indicated by the switch in color from red (Beefmaster genotype) to yellow (Sungold genotype). The mean fruit weight for the congenic lines carrying these recombinant chromosomes is indicated on the right. By inspection of Figure 19-16, you’ll notice that all lines with the Beefmaster allele of kin1 (a kinase gene) have fruit of ∼180 g, while those with the Sungold allele of kin1 have fruit of about ∼170 g. None of the other genes are associated with fruit weight in this way. If confirmed by appropriate statistical tests, this result allows us to identify kin1 as the gene underlying this QTL.
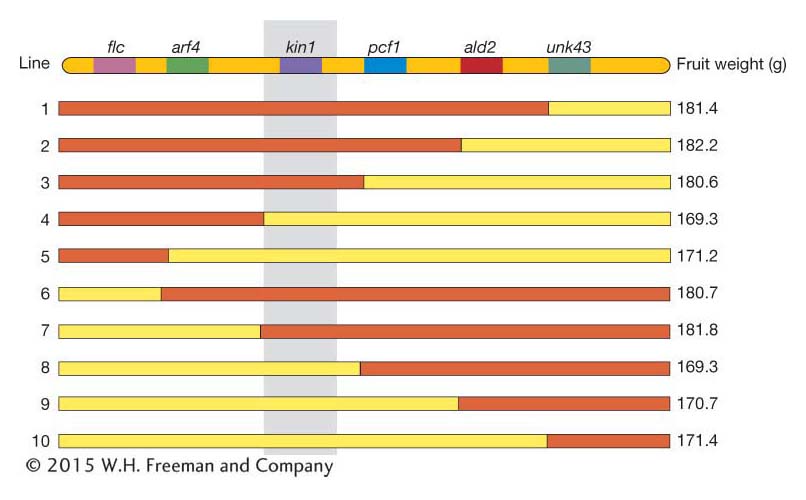
Table 19-7 lists a small sample of the hundreds of genes or QTL affecting quantitative variation from different species that have been identified. The list includes the gene for maize flowering time, Vgt, that underlies one of the Lod peaks in Figure 19-15a. One notable aspect of this list is the diversity of gene functions. There does not appear to be a rule that only particular types of genes can be a QTL. Most, if not all, genes in the genomes of organisms are likely to contribute to quantitative variation in populations.
|
Organism |
Trait |
Gene |
Gene function |
|---|---|---|---|
|
Yeast |
High- |
RHO2 |
GTPase |
|
Arabidopsis |
Flowering time |
CRY2 |
Cryptochrome |
|
Maize |
Branching |
Tb1 |
Transcription factor |
|
Maize |
Flowering time |
Vgt |
Transcription factor |
|
Rice |
Photoperiod sensitivity |
Hd1 |
Transcription factor |
|
Rice |
Photoperiod sensitivity |
CK2a |
Casein kinase a subunit |
|
Tomato |
Fruit- |
Brix9- |
Invertase |
|
Tomato |
Fruit weight |
Fw2.2 |
Cell- |
|
Drosophila |
Bristle number |
Scabrous |
Secreted glycoprotein |
|
Cattle |
Milk yield |
DGAT1 |
Diacylglycerol acyltransferase |
|
Mice |
Colon cancer |
Moml |
Modifier of a tumor- |
|
Mice |
Type 1 diabetes |
I- |
Histocompatibility antigen |
|
Humans |
Asthma |
ADAM33 |
Metalloproteinase- |
|
Humans |
Alzheimer’s disease |
ApoE |
Apolipoprotein |
|
Humans |
Type 1 diabetes |
HLA- |
MHC class II surface glycoprotein |
|
Source: A. M. Glazier et al., Science 298, 2002, 2345– |
|||
KEY CONCEPT
Quantitative trait locus (QTL) mapping is a procedure for identifying the genomic locations of the genes (QTL) that control variation for quantitative or complex traits. QTL mapping evaluates the progeny of controlled crosses for their genotypes at molecular markers and for their trait values. If the different genotypes at a marker locus have different mean values for the trait, then there is evidence for a QTL near the marker. Once a region of the genome containing a QTL has been identified, QTL can be mapped to single genes using congenic lines.749