20.6 The Origin of New Genes and Protein Functions
Evolution consists of more than the substitution of one allele for another at loci of defined function. A large fraction of protein-
787
Expanding gene number
There are several genetic mechanisms that can expand the number of genes or parts of genes. One large-
A second mechanism that can increase gene number is gene duplication. Misreplication of DNA during meiosis can cause segments of DNA to be duplicated. The lengths of the segments duplicated can range from just one or two nucleotides up to substantial segments of chromosomes containing scores or even hundreds of genes. Detailed analyses of human-
A third mechanism that can generate gene duplications is transposition. Sometimes, when a transposable element is transposed to another part of the genome, it may carry along additional host genetic material and insert a copy of some part of the genome into another location (see Chapter 15).
788
A fourth mechanism that can expand gene number is retrotransposition. Many animal genomes harbor retroviral-
The fate of duplicated genes
It was once thought that because the ancestral function is provided by the original gene, duplicate genes are essentially spare genetic elements that are free to evolve new functions (termed neofunctionalization), and that would be a common fate. However, the detailed analysis of genomes and population-
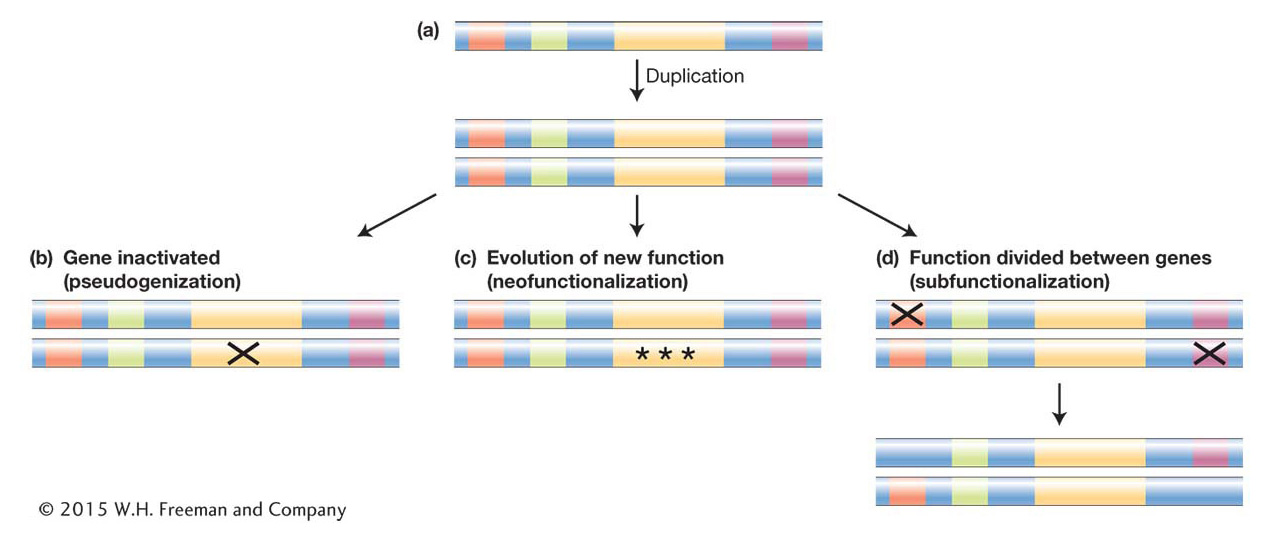
For simplicity’s sake, let’s consider a duplication event that results in the duplication of the entire coding and regulatory region of a gene (Figure 20-19a). Many different outcomes can unfold from such a duplication. The simplest result is that the allele bearing the duplicate is lost from the population before it rises to any significant frequency, as is the fate of many new mutations (see Chapter 18). But let’s consider next the more interesting scenarios: suppose the duplication survives and new mutations begin to occur within the duplicate gene pair. Keeping in mind that the original and duplicated genes are initially exact copies and therefore redundant, once new mutations arise, there are several possible fates:
An inactivating mutation may occur in the coding region of either duplicate. The inactivated paralog is called a pseudogene and will generally be invisible to natural selection. Thus, it will accumulate more mutations and evolve by random genetic drift, while natural selection will maintain the functional paralog (Figure 20-19b).
Mutations may occur that alter the regulation of one duplicate or the activity of one encoded protein. These alleles may then become subject to positive selection and acquire a new function (neofunctionalization) (Figure 20-19c).
In cases where the ancestral gene has more than one function and more than one regulatory element, as for most toolkit genes, a third possible outcome is that initial mutations inactivate or alter one regulatory element in each duplicate. The original gene function is now divided between the duplicates, which complement each other. In order to preserve the ancestral function, natural selection will maintain the integrity of both gene-
coding regions. Loci that follow this path of duplication and mutation that produce complementary paralogs are said to be subfunctionalized (Figure 20-19d).
Some of these alternative fates of gene duplicates are illustrated in the history of the evolution of human globin genes. The evolution of our lineage, from fish ancestors to terrestrial amniotes that laid eggs to placental mammals, has required a series of innovations in tissue oxygenation. These include the evolution of additional globin genes with novel patterns of regulation and the evolution of hemoglobin proteins with distinct oxygen-
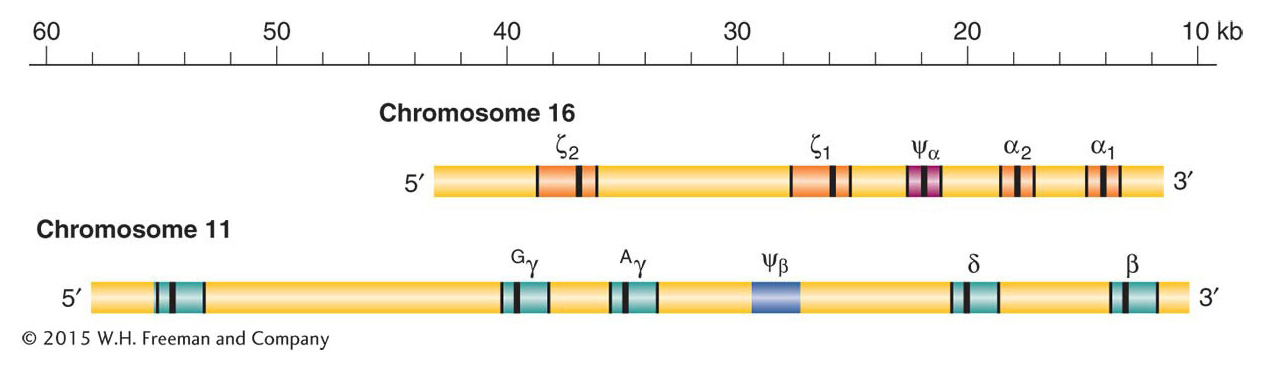
Adult hemoglobin is a tetramer consisting of two α polypeptide chains and two β chains, each with its bound heme molecule. The gene encoding the adult α chain is on chromosome 16, and the gene encoding the β chain is on chromosome 11. The two chains are about 49 percent identical in their amino acid sequences; this similarity reflects their common origin from an ancestral globin gene deep in evolutionary time. The α chain gene resides in a cluster of five related genes (α and ζ) on chromosome 16, while the β chain resides in a cluster of six related genes on chromosome 11 (ε, β, δ, and γ) (Figure 20-20). Each cluster contains a pseudogene, Ψα and Ψβ, respectively, that has accumulated random, inactivating mutations.
789
Each cluster contains genes that have evolved distinct expression profiles, a distinct function, or both. Of greatest interest are the two γ genes. These genes are expressed during the last seven months of fetal development to produce fetal hemoglobin (also known as hemoglobin F), which is composed of two α chains and two γ chains. Fetal hemoglobin has a greater affinity for oxygen than does adult hemoglobin, which allows the fetus to extract oxygen from the mother’s circulation via the placenta. At birth, up to 95 percent of hemoglobin is the fetal type, then expression of the adult β form replaces γ and a small amount of δ globin is also produced. The order of appearance of globin chains during development is orchestrated by a complex set of cis-
790
The γ genes are restricted to placental mammals. Their distinct developmental regulation and protein products mean that these duplicates have evolved differences in function that have contributed to the evolution of the placental lifestyle. Interestingly, regulatory variants of these genes are known that cause expression of the fetal hemoglobin to persist into childhood and adulthood. These naturally occurring variants appear to moderate the severity of sickle-