Observations in humans led to the proposal that genes determine enzymes
The identification of a gene product as a protein began with a mutation. In the early twentieth century, English physician Archibald Garrod saw several children with a rare disease. One symptom of the disease was that the urine turned dark brown when exposed to air, which was especially noticeable on the infants’ diapers. The disease was given the descriptive name alkaptonuria (“black urine”).
Garrod noticed that the disease was most common in children whose parents were first cousins. Mendelian genetics had just been “rediscovered,” and Garrod realized that because first cousins share on average 1⁄8 of their alleles, the children of first cousins might inherit a rare mutant allele from both parents. He proposed that alkaptonuria was a recessive phenotype caused by a mutant allele that causes the disease phenotype.
Garrod took the analysis further by identifying the biochemical abnormality in the affected children. He isolated from them an unusual substance, homogentisic acid, which accumulated in blood, joints (where it crystallized and caused severe pain), and urine (where it turned black). The chemical structure of homogentisic acid is similar to that of the amino acid tyrosine:
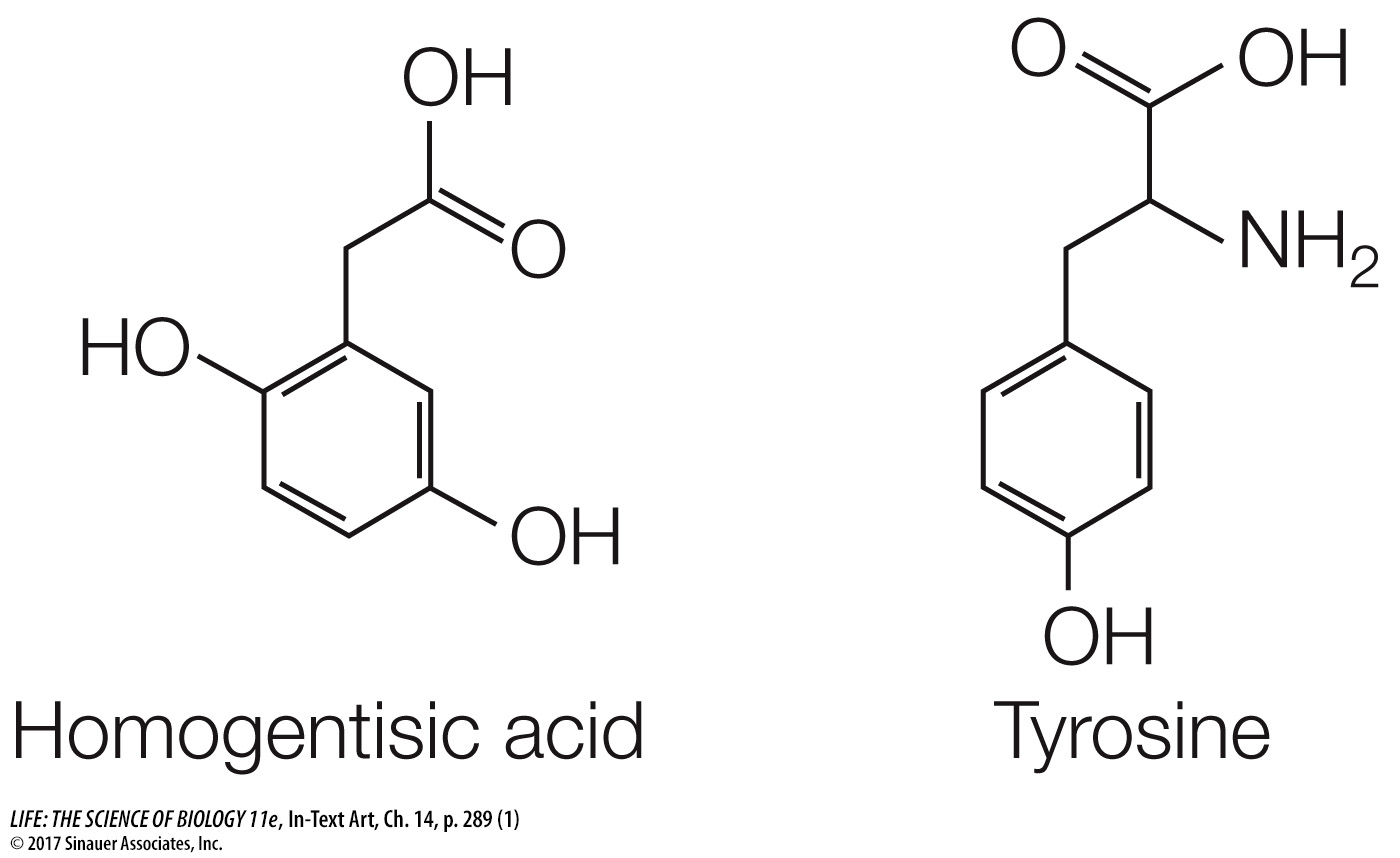
The function of enzymes as biological catalysts had just been discovered. Garrod proposed that homogentisic acid was a breakdown product of tyrosine. He suggested that while homogentisic acid ordinarily would be converted to a harmless product, it was not being converted in children with alkaptonuria. Garrod further hypothesized that the enzyme required for the breakdown of homogentisic acid was not being produced in these children. He suggested that a normal human allele (for the wild-
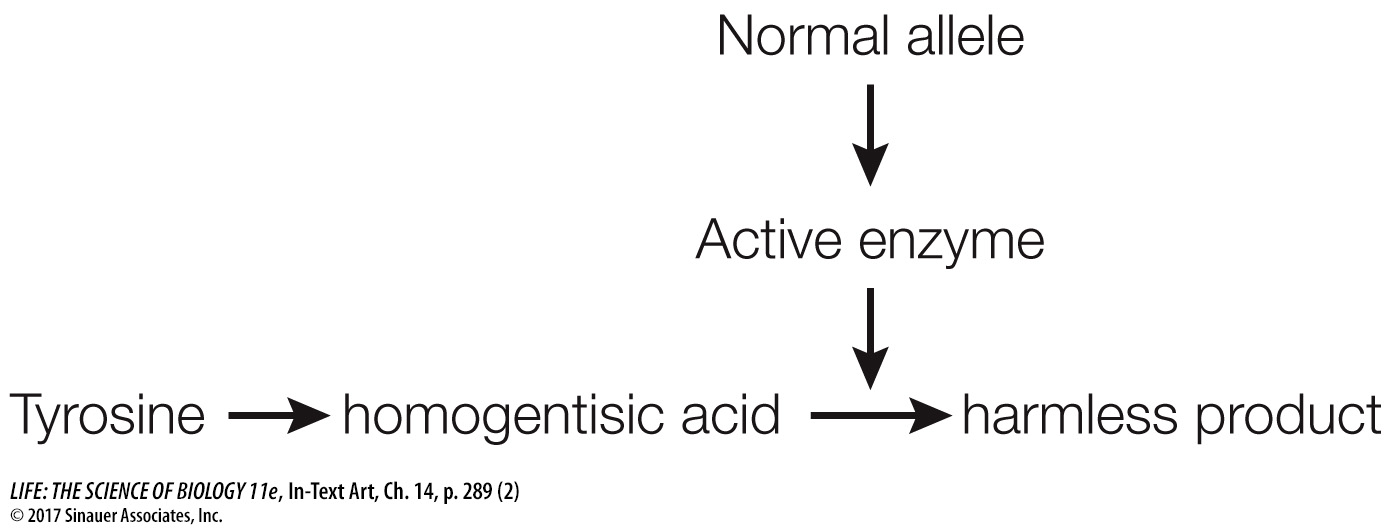
If the allele were mutated, the enzyme would be inactive and homogentisic acid would accumulate instead:
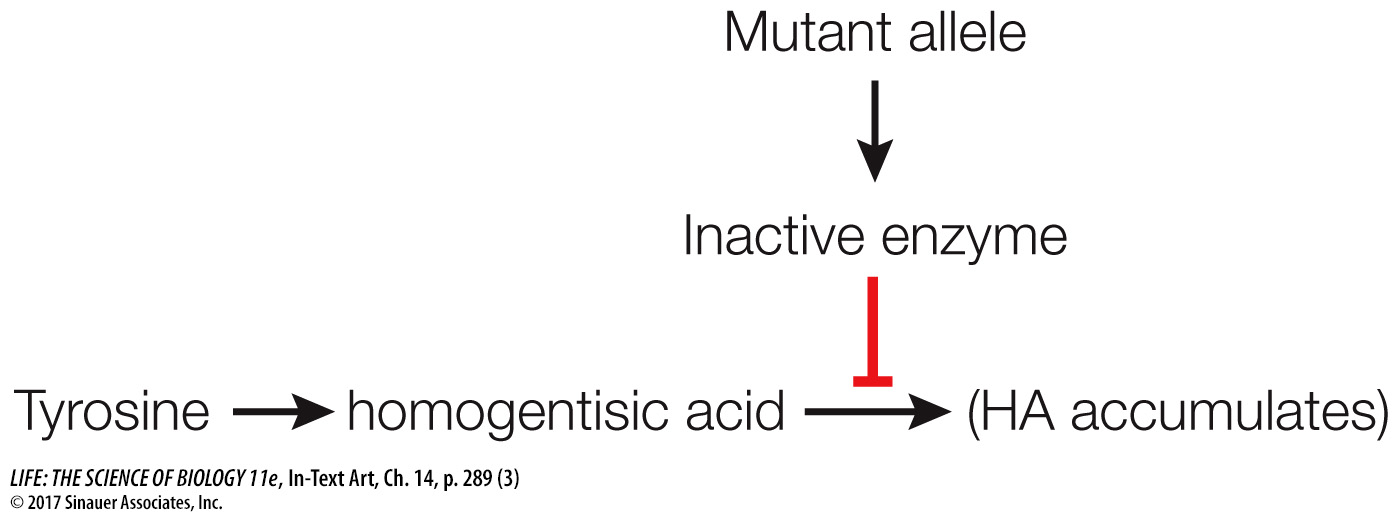
Garrod concluded that there must be one gene to one enzyme and coined the term “inborn error of metabolism” to describe this genetically determined biochemical disease. But his hypothesis was not confirmed until the actual enzyme and the specific gene mutation involved were identified. The enzyme, homogentisic acid oxidase, was identified as active in healthy people and inactive in alkaptonuria patients in 1958, and the specific DNA mutation was described in 1996.
To relate genes and enzymes more generally, biologists turned to simpler organisms that could be manipulated in the laboratory.