Disease-causing mutations may make proteins dysfunctional
Genetic mutations are often expressed phenotypically as proteins that differ from normal (wild-type) proteins. Abnormalities in enzymes, receptor proteins, transport proteins, structural proteins, and most of the other functional classes of proteins have all been implicated in genetic diseases.
LOSS OF ENZYME FUNCTION In 1934, the urine of two intellectually disabled young siblings was found to contain phenylpyruvic acid, an unusual by-product of the metabolism of the amino acid phenylalanine. It took two decades for scientists to trace the complex clinical phenotype of the disease that afflicted these children, called phenylketonuria (PKU), back to its molecular cause. The disease results from an abnormality in a single enzyme, phenylalanine hydroxylase (PAH), which catalyzes the conversion of dietary phenylalanine to tyrosine (Figure 15.6). This enzyme is not active in the livers of PKU patients, leading to excesses of phenylalanine and phenylpyruvic acid in the blood. Since then, the nucleotide sequence of the PAH gene has been compared between healthy people and those with the PKU disease, and more than 400 different disease-causing mutations have been found. The most common one is a missense mutation that results in tryptophan instead of arginine at position 408 in the polypeptide chain (Table 15.1). As is often the case with loss-of-function mutations, the mutant alleles are recessive, because one functional allele is all that is needed to produce enough functional PAH to prevent the disease.
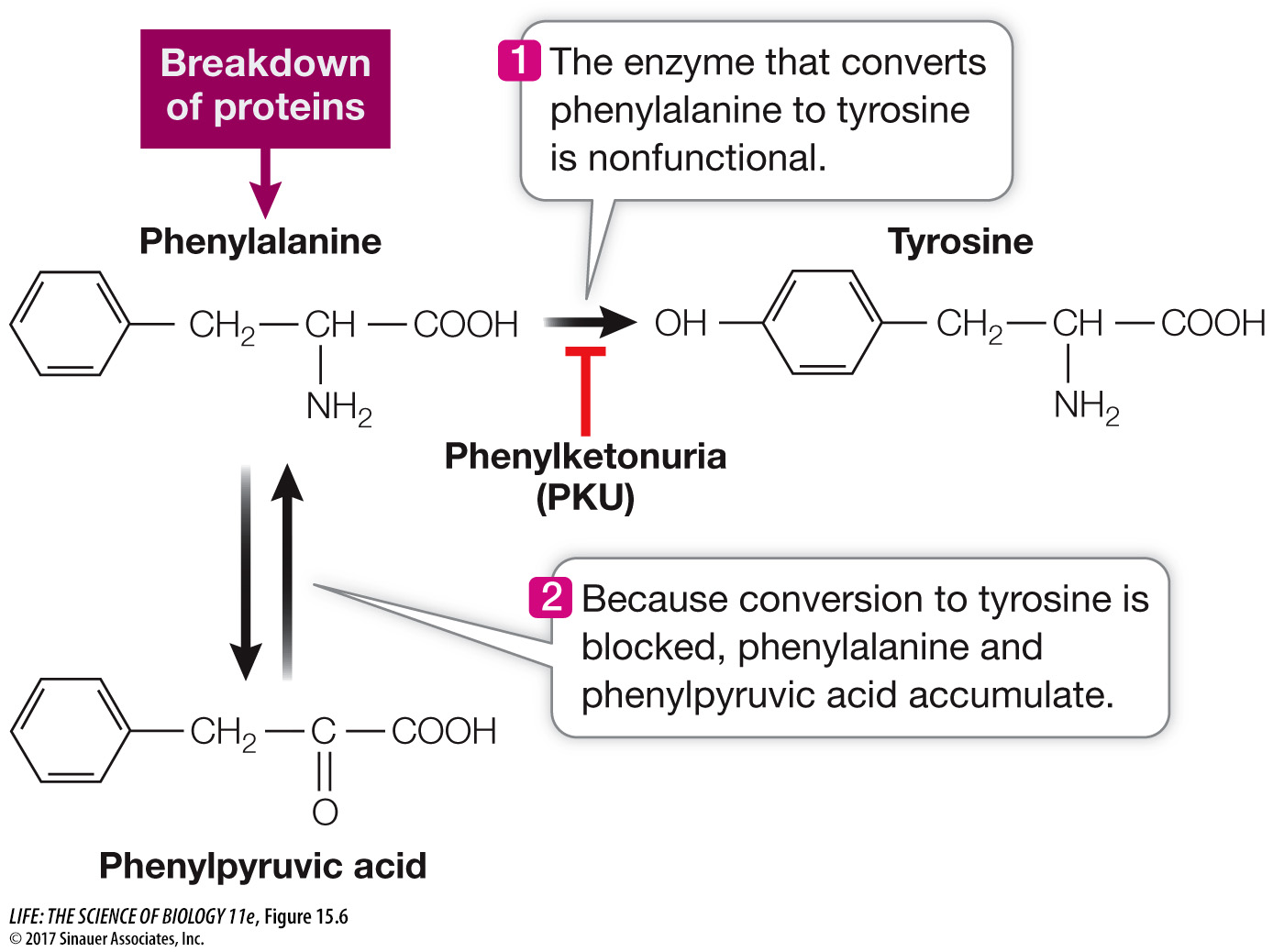
Figure 15.6 One Gene, One Enzyme Phenylketonuria is caused by an abnormality in a specific enzyme that metabolizes the amino acid phenylalanine. Knowing the molecular causes of such single-gene, single-enzyme metabolic diseases can aid researchers in developing screening tests as well as treatments.
table 15.1 Two Common Mutations That Cause Phenylketonuria
|
Codon 408 (20% of PKU cases) |
|
Codon 280 (2% of PKU cases) |
|
Normal |
Mutant |
|
Normal |
Mutant |
| Length of PAH protein |
452 amino acids |
452 amino acids |
|
452 amino acids |
452 amino acids |
| DNA at codon |
...CGG... |
...TGG... |
|
...GAA... |
...AAA... |
|
...GCC... |
...ACC... |
|
...CTT... |
...TTT... |
| mRNA at codon |
...CGG... |
...UGG... |
|
...GAA... |
...AAA... |
| Amino acid at codon |
Arginine |
Tryptophan |
|
Glutamic acid |
Lysine |
| Active PAH enzyme? |
Yes |
No |
|
Yes |
No |
Hundreds of human genetic diseases that result from enzyme abnormalities have been discovered, some of which lead to intellectual disability and premature death. Most of these diseases are rare; PKU, for example, shows up in 1 out of every 12,000 newborns. As you by now realize, for any given gene there may be numerous alleles. Some alleles encode proteins that function normally, whereas others produce proteins that cause disease. Let’s take a look at a gene whose alleles illustrate both of these circumstances—the gene for one of the polypeptide chains in hemoglobin.
ABNORMAL HEMOGLOBIN As mentioned in Key Concept 15.1, sickle-cell disease is caused by a recessive, missense mutation. This blood disorder most often afflicts people whose ancestors came from the tropics or from the Mediterranean region.
Recall that human hemoglobin is composed of four globin subunits—two α-chains and two β-chains—as well as the pigment heme (see Figure 3.11). In sickle-cell disease, one of the 146 amino acids in the β-globin polypeptide chain is abnormal: at position 6, glutamic acid is replaced by valine. This replacement changes the charge of the protein (glutamic acid is hydrophilic and valine is hydrophobic), causing it to form long, needlelike aggregates in the red blood cells. The phenotypic result is sickle-shaped red blood cells and an impaired ability of the blood to carry oxygen. The sickled cells tend to block narrow blood capillaries, resulting in tissue damage and eventually death by organ failure.
Because hemoglobin is easy to isolate and study, its variations in the human population have been extensively documented (Figure 15.7). Hundreds of single amino acid alterations in β-globin, all due to mutations forming alleles, have been reported. For example, at the same position that is mutated in sickle-cell disease (resulting in hemoglobin S), glutamic acid may be replaced by lysine, causing hemoglobin C disease. In this case, the resulting anemia is usually not severe. Many alleles that result in alterations of the amino acid sequence of hemoglobin do not affect its function. In fact, about 5 percent of us carry at least one missense point mutation in a β-globin allele.
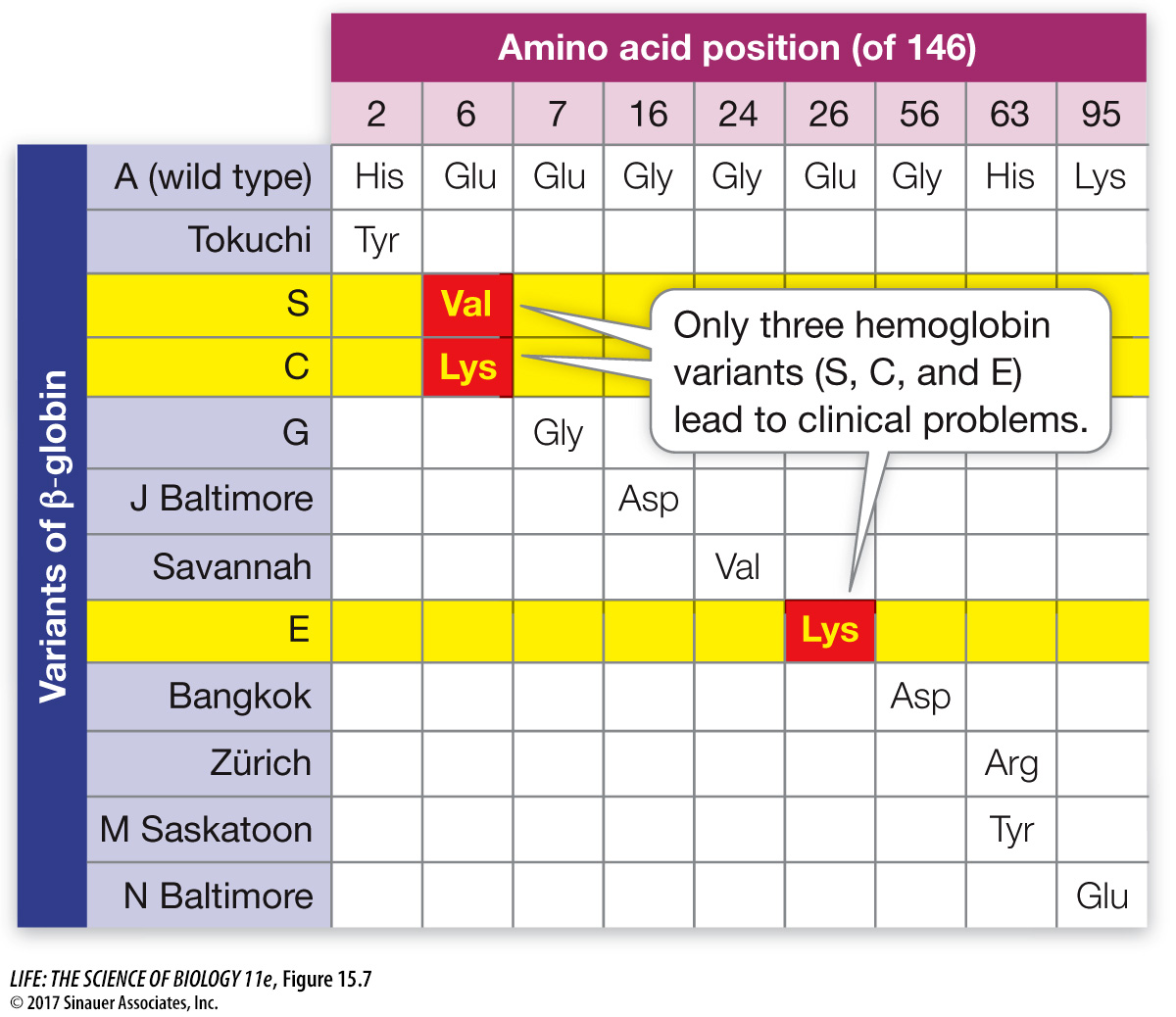
Figure 15.7 Hemoglobin Polymorphism Each of these mutant alleles codes for a protein with a single amino acid change in the 146-amino acid chain of β-globin. Only three of the hundreds of known variants of β-globin, shown on the left, are known to lead to clinical abnormalities. “S” is the sickle-cell anemia allele.
Some of the more common examples of inherited diseases caused by specific protein defects are listed in Table 15.2. These mutations can be dominant, codominant, or recessive, and some are sex-linked.
table 15.2 Some Human Genetic Diseases
| Disease name |
Inheritance pattern; births frequency |
Gene mutated; protein product |
Clinical phenotype |
| Familial hypercholesterolemia |
Autosomal codominant; 1 in 500 heterozygous |
LDLR; low-density lipoprotein receptor |
High blood cholesterol, heart disease |
| Cystic fibrosis |
Autosomal recessive;
1 in 4,000 |
CFTR; chloride ion channel in membrane |
Immune, digestive, and respiratory illness |
| Duchenne muscular dystrophy |
Sex-linked recessive;
1 in 3,500 males |
DMD; the muscle membrane protein dystrophin |
Muscle weakness |
| Hemophilia A |
Sex-linked recessive;
1 in 5,000 males |
HEMA; factor VIII blood clotting protein |
Inability to clot blood after injury, hemorrhage |