The base sequence of a short DNA fragment can be determined quickly
The notion of sequencing the entire genome of a complex organism was not contemplated until 1986. The Nobel laureate Renato Dulbecco and others proposed at that time that the world scientific community be mobilized to undertake the sequencing of the entire human genome. One motive was to detect DNA damage in people who had survived the atomic bomb attacks and been exposed to radiation in Japan during World War II. But in order to detect changes in the human genome, scientists first needed to know its sequence.
The result was the publicly funded Human Genome Project, an enormous undertaking that was successfully completed in 2003. This effort was aided and complemented by privately funded groups. The genome project benefited from the development of many new methods that were first developed to sequence smaller genomes—
Many prokaryotes have a single chromosome, whereas eukaryotes have many. Because of their differing sizes, chromosomes are easy to separate from one another. You might think that the straightforward way to sequence a chromosome would be to start at one end and simply sequence the DNA molecule one nucleotide at a time. The task is somewhat simplified because only one of the two strands needs to be sequenced, the other being complementary. So if you have the sequence
5′ AAGCTCA.....3′
you know that the other strand must be
3′ TTCGAGT.....5′.
But sequencing a DNA molecule millions of bp long from one end to the other is just not possible using current methods. At most, about a few thousand bp can be sequenced at a time using current methods. The key to determining genome sequences is to break up long chromosomes into smaller DNA fragments and then thousands of fragments are sequenced individually at the same time.
In the 1970s Frederick Sanger and his colleagues invented a way to sequence DNA by using chemically modified nucleotides that were originally developed to stop cell division in cancer. This method, or a variation of it, was used to obtain the first human genome sequence as well as those of several model organisms. However, it was relatively slow, expensive, and labor-
*connect the concepts PCR can be automated and is a key to sequencing small amounts of DNA. Learn about PCR in Key Concept 13.5.
High-
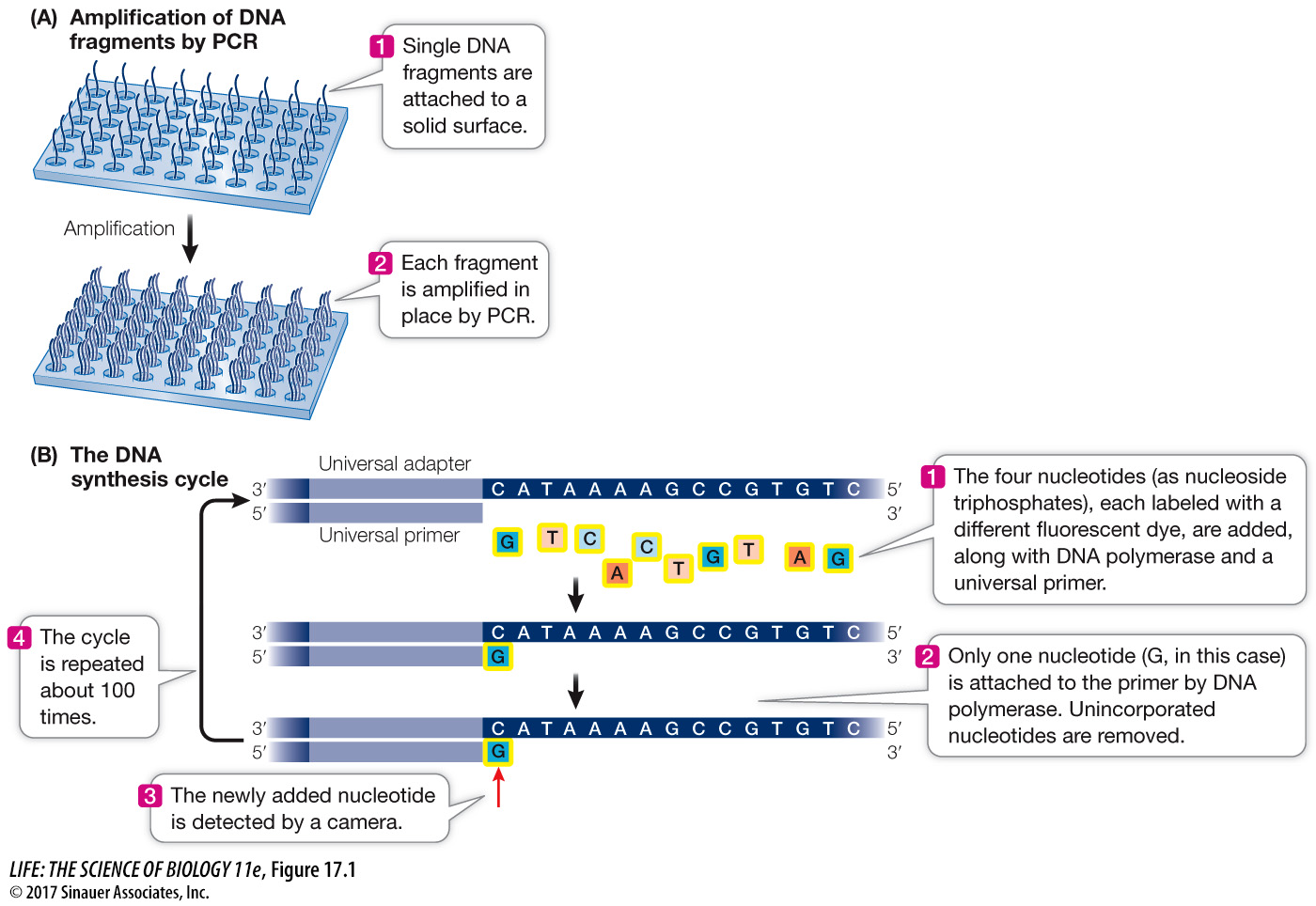
A large molecule of DNA is cut into small fragments of about 100 bp each. This can be done physically, using mechanical forces to shear (break up) the DNA, or by using enzymes that hydrolyze the phosphodiester bonds between nucleotides at intervals in the DNA backbone.
The DNA is denatured by heat, breaking the hydrogen bonds that hold the two strands together. Each single strand acts as a template for the synthesis of new, complementary DNA.
Short, synthetic oligonucleotides are attached to each end of each fragment, and these are attached to a solid support.
The DNA is amplified by PCR using primers complementary to the synthetic oligonucleotides attached to the ends of each DNA. The multiple (approximately 1,000) copies of the DNA at a single location allow for easy detection of added nucleotides during the sequencing steps.
Animation 17.1 Sequencing the Genome
www.life11e.com/
Animation 17.2 High-
www.life11e.com/
361
Once the DNA has been attached to a solid substrate and amplified, it is ready for sequencing (see Figure 17.1B):
At the beginning of each sequencing cycle, the fragments are heated to denature them. A solution containing a universal primer (complementary to one of the same synthetic oligonucleotides used for the PCR amplification step), DNA polymerase, and the four deoxyribonucleoside triphosphates (dNTPs: dATP, dGTP, dCTP, and dTTP) is then added to the DNA. Recall that dNTPs are the substrates that the DNA polymerase uses in DNA synthesis (see Key Concept 13.3). Each of the four kinds of dNTP is tagged with a different colored fluorescent dye.
The DNA synthesis reaction is set up so that only one nucleotide is added to the new DNA strand in each sequencing cycle. After each addition, the unincorporated dNTPs are removed.
The fluorescence of the new nucleotide at each location is detected with a camera. The color of the fluorescence indicates which of the four nucleotides was added.
The fluorescent tag is removed from the nucleotide that is already attached, and then the DNA synthesis cycle is repeated. Images are captured after each nucleotide is added. The series of colors at each location indicate the sequence of nucleotides in the growing DNA strand at that location.
The power of this method derives from the fact that:
It is fully automated and miniaturized.
Millions of different fragments are sequenced at the same time.
It is an inexpensive way to sequence large genomes. For example, at the time of this writing, a complete human genome could be sequenced in less than a day for $1,000. This is in contrast to the Human Genome Project, which took 13 years and $2.7 billion to sequence one genome!
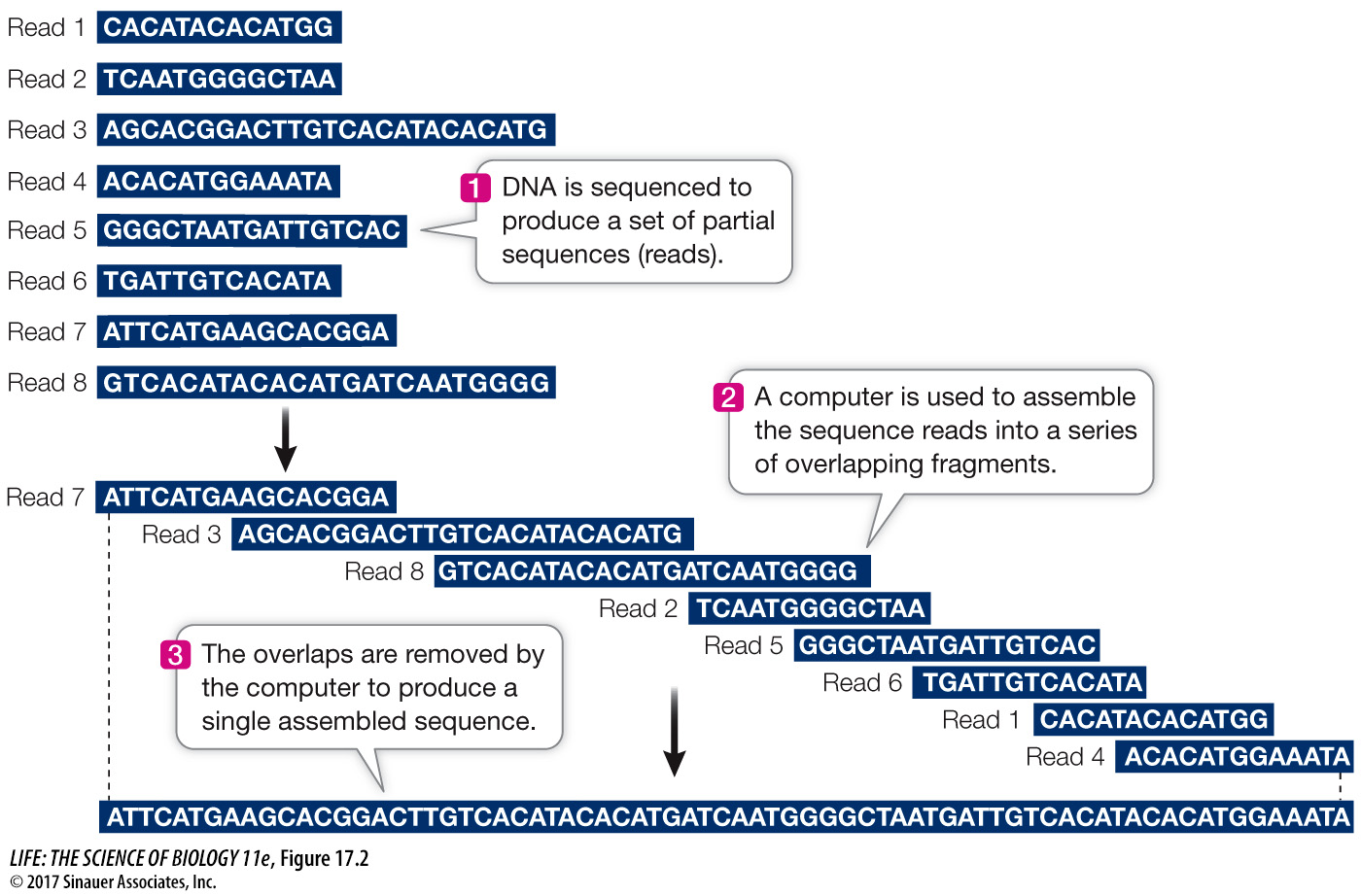
Sequencing millions of short DNA fragments is only part of the process of constructing the genome. Once these sequences have been determined, they need to be put together in the correct order. That is, they need to be arranged to reflect the sequence of the chromosomes from which they came. Imagine if you cut out every word in this book (there are more than half a million of them), put them on a table, and tried to arrange them in their original order! The enormous task of determining DNA sequences is possible because the original DNA fragments are overlapping.
Let’s illustrate the process using a single 10-
TG, ATG, and CCTAC
362
Cutting the same molecule with the second enzyme generates the fragments:
AT, GCC, and TACTG
Cutting with the third enzyme results in:
CTG, CTA, and ATGC
Can you put the fragments in the correct order? (The answer is ATGCCTACTG.) For genome sequencing, the sequence fragments are called “reads” (Figure 17.2). Of course, the problem of ordering 2.5 million fragments from human chromosome 1 (246 million bp) is more challenging than our 10-