Molecular evolution is used to study and combat diseases
Many of the most problematic human diseases are caused by living, evolving organisms that present a moving target for modern medicine. Recall the example of influenza described in the opening story of this chapter and that of HIV in Chapter 21. The control of these and many other human diseases depends on techniques that can track the evolution of pathogenic organisms over time.
The transportation advances of the past century have allowed humans to move around the world with unprecedented speed and frequency. Unfortunately, this mobility has allowed pathogens to be transmitted among human populations, and between humans and other animals, at increasing rates. Cross-
In the opening of this chapter, we described the devastating influenza pandemic of 1918–
Studies of the genomes of many infectious agents have advanced our understanding and treatment of the diseases they cause. For example, rodent-
503
investigating life
Why Was the 1918–
experiment
Original Papers: Tumpey, T. M. et al. 2005. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science 310: 77–
Worobey, M., G.-Z. Han, and A. Rambaut. 2014. A synchronized global sweep of the internal genes of modern avian influenza virus. Nature 508: 254–
As conveyed in the story that opens this chapter, the 1918–
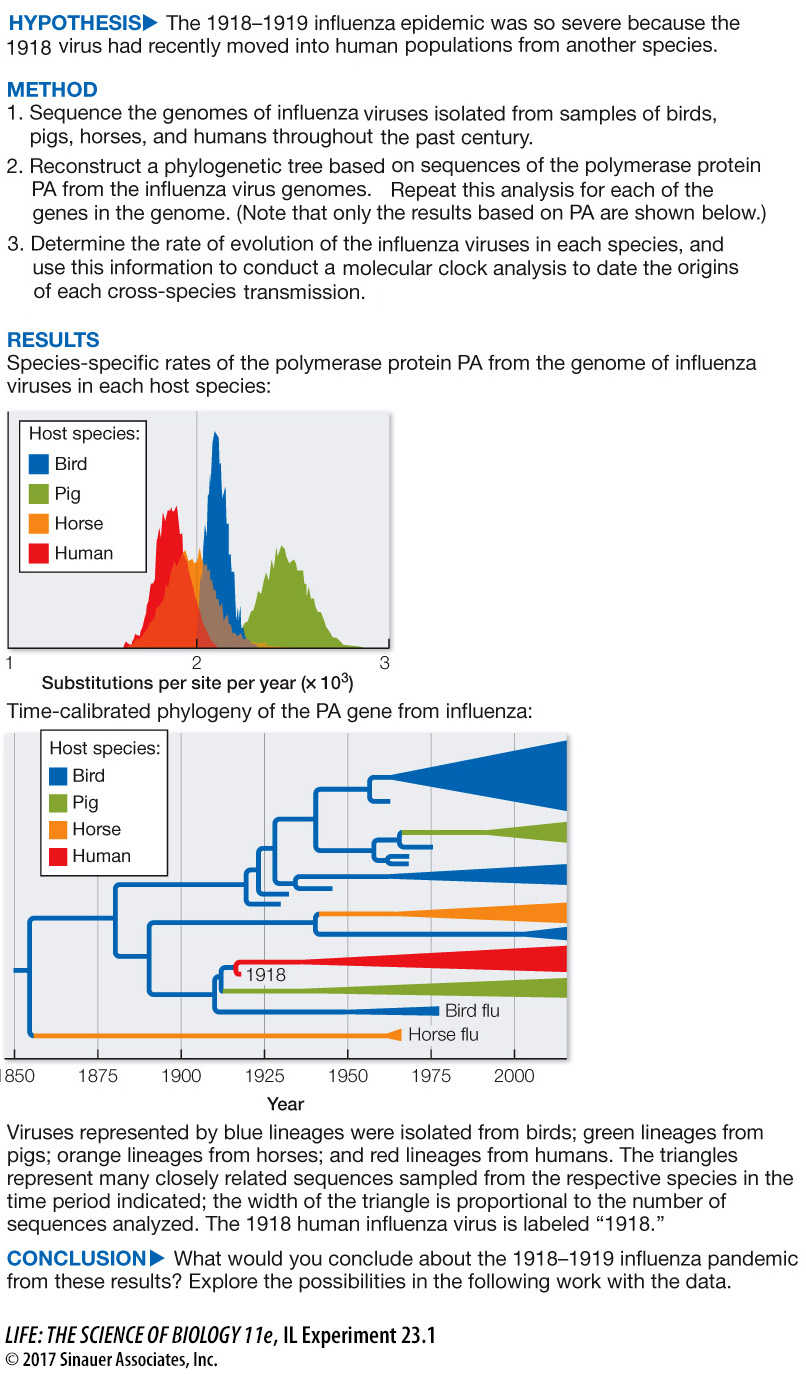
Terrence Tumpey and his colleagues at the Centers for Disease Control and Prevention isolated and sequenced a complete flu virus from biopsies of 1918 victims of the pandemic. Michael Worobey of the University of Arizona and his colleagues then analyzed and compared the genomes of flu viruses that have been collected from 1918 to the present, from birds, pigs, horses, and humans. The viruses are largely transmitted within each of these host groups, but rarely jump between host groups. Worobey and his colleagues reconstructed the phylogeny of the viruses to determine if the 1918 virus had recently “jumped” into human populations from another species.
work with the data
Original Paper: Worobey, M., G.-Z. Han and A. Rambaut. 2014.
Michael Worobey and his colleagues were able to reconstruct many of the cross-
QUESTIONS
Question 1
All the influenza viruses shown in the tree in the experiment are thought to have arisen in one group of host animals. Which group of animals do you think is the original source of the virus, and why?
Birds appear to be the original source of influenza viruses that now cause flu in the various mammal species (horses, pigs, and humans). The bird lineages extend to the base of the tree, and appear to be closely related to each lineage of influenza virus that occurs in a mammal.
Question 2
How many cross-
The tree shows five cross-
Question 3
What do you think is a likely explanation for the severity of the 1918–
In 1918, a new strain of influenza had just entered human populations (as well as pig populations). Humans' immune systems would not have had any prior experience with this new strain of influenza, and so would not have been prepared for an effective defense.
Question 4
Influenza was known in humans well before the 1918–
Earlier flu cases were likely caused by other cross-
A similar work with the data exercise may be assigned in LaunchPad.
504
In the future, molecular evolution will become even more critical to the identification of human (and other) diseases. Once biologists have collected data on the genomes of enough organisms, it will be possible to identify an infection by sequencing a portion of the infecting organism’s genome and comparing this sequence with other sequences on an evolutionary tree. At present it is difficult to identify many common viral infections (those that cause “colds,” for instance). As genomic databases and evolutionary trees increase, however, automated methods of sequencing and rapid phylogenetic comparison of the sequences will allow us to identify and treat a much wider array of human illnesses.