Genes and proteins are compared through sequence alignment
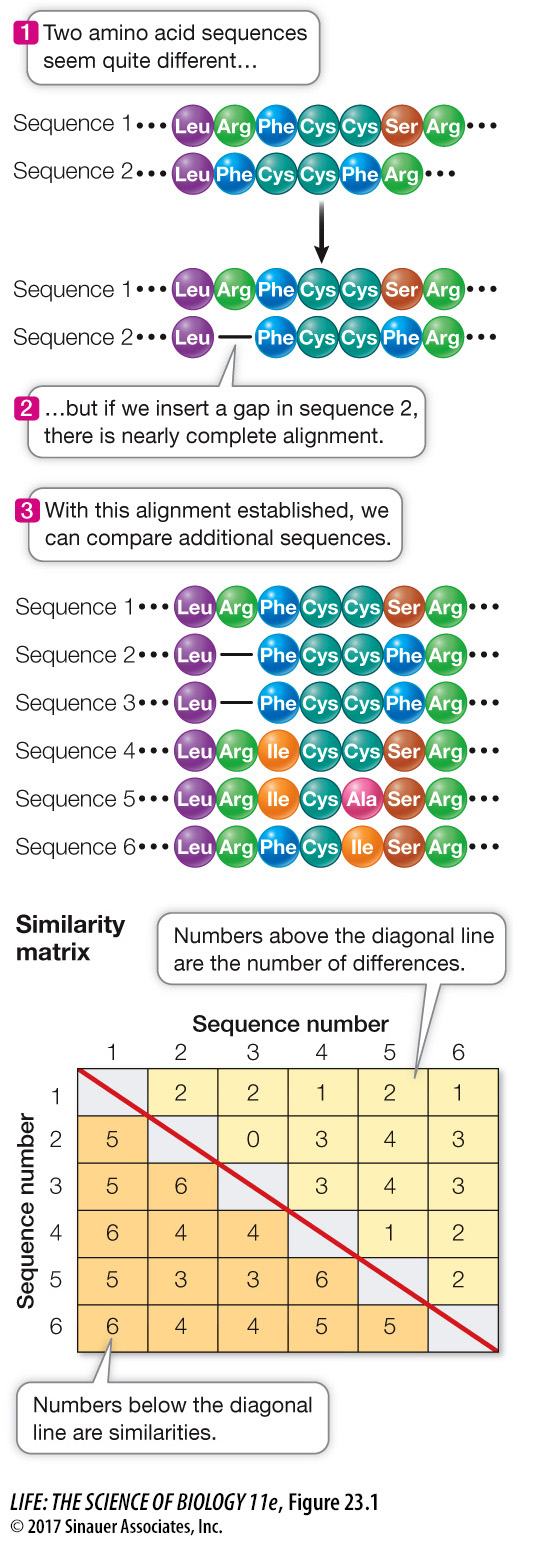
Once the nucleotide or amino acid sequences of molecules from different organisms have been determined, they can be compared. Homologous positions can be identified only if we first pinpoint the locations of deletions and insertions that have occurred in the molecules of interest in the time since the organisms diverged from a common ancestor. A simple hypothetical example illustrates this sequence alignment technique. In Figure 23.1 we compare two amino acid sequences from homologous proteins in different organisms. The two sequences at first appear to differ in both the number and identity of their amino acids. If we insert a gap after the first amino acid in sequence 2 (after leucine), however, the similarities in the two sequences become obvious. This gap represents the occurrence of one of two evolutionary events: an insertion of an amino acid in the longer protein or a deletion of an amino acid in the shorter protein. Having adjusted for this insertion or deletion event, we can see that the two sequences differ by only one amino acid at position 6 (serine or phenylalanine).
488
research tools
Figure 23.1 Amino Acid Sequence Alignment
Amino acid sequence alignment is a way of arranging protein sequences to identify regions of homology between the sequences. Gaps are inserted between the amino acid residues to align similar residues in columns. Differences (number of amino acid differences plus insertion or deletion events) and similarities (number of identical amino acids) between each pair of aligned sequences are then summarized in a similarity matrix. Homologous DNA sequences can be aligned in a similar manner.
Activity 23.1 Amino Acid Sequence Alignment
www.life11e.com/
Adding a single gap—
Having aligned the sequences, we can compare them by counting the number of nucleotides or amino acids that differ between them. Summing the numbers of the same and different amino acids in each pair of sequences allows us to construct a similarity matrix, which gives us a measure of the minimum number of changes that have occurred since the divergence of each pair of organisms (see Figure 23.1).
Activity 23.2 Similarity Matrix Construction
www.life11e.com/