Chapter 21
RECAP 21.1
Phylogenetic trees can represent any process in which biological lineages diverge, such as speciation (in which case the tree will depict the evolutionary relationships among species), viral replication (in which case the tree will depict the evolutionary relationships among different viral lineages), or gene duplication (in which case the tree will depict the evolutionary relationships among genes). In most cases in this book, phylogenetic trees are used to depict the evolutionary relationships among species or higher groups of organisms.
Page A-22Because homologous characters are similar as a result of their common descent. Similarities that result from convergent evolution (the wings of birds and insects, for example) can be misleading about evolutionary relationships if they are mistaken as homologous characters.
Selection for similar environmental conditions often leads to convergence in traits. For example, fish and dolphins both have fins and are similar in body shape because there is strong selection for these traits in an aquatic environment. But these traits have evolved independently in the two groups. Biologists can usually detect such homoplasies because they conflict with a large number of homologous traits in the groups that are similar as a result of their recent shared ancestry. Biologists can minimize homoplasies using the principle of parsimony.
RECAP 21.2
The pine tree and the sunflower.
The fern.
True roots and vascular cells.
No, because each trait can be placed along a single branch in the tree.
RECAP 21.3
The West Nile Virus in the United States appears to be most closely related to a strain of the virus isolated in Israel. A reasonable hypothesis is that the virus emerged in Africa in the 1930s and subsequently moved into Asia and Europe, probably multiple times. In the late 1990s, a strain of the virus from Israel appears to have been transported to New York, perhaps carried by mosquitoes on an airplane or in a cargo shipment. Once in the United States, the virus spread quickly in native bird populations across North America.
The rate of divergence is represented by the slope of the regression line. Since the regression line goes approximately through the origin (0, 0), the slope is approximately 7.5% change in cyt b per 4 my, or 1.875% change per million years.
RECAP 21.4
Classifications One and Three (the group Reptilia is paraphyletic if the birds are excluded).
Classification Three (the group Homotheria is polyphyletic).
Classification Two.
WORK WITH THE DATA, P. 457
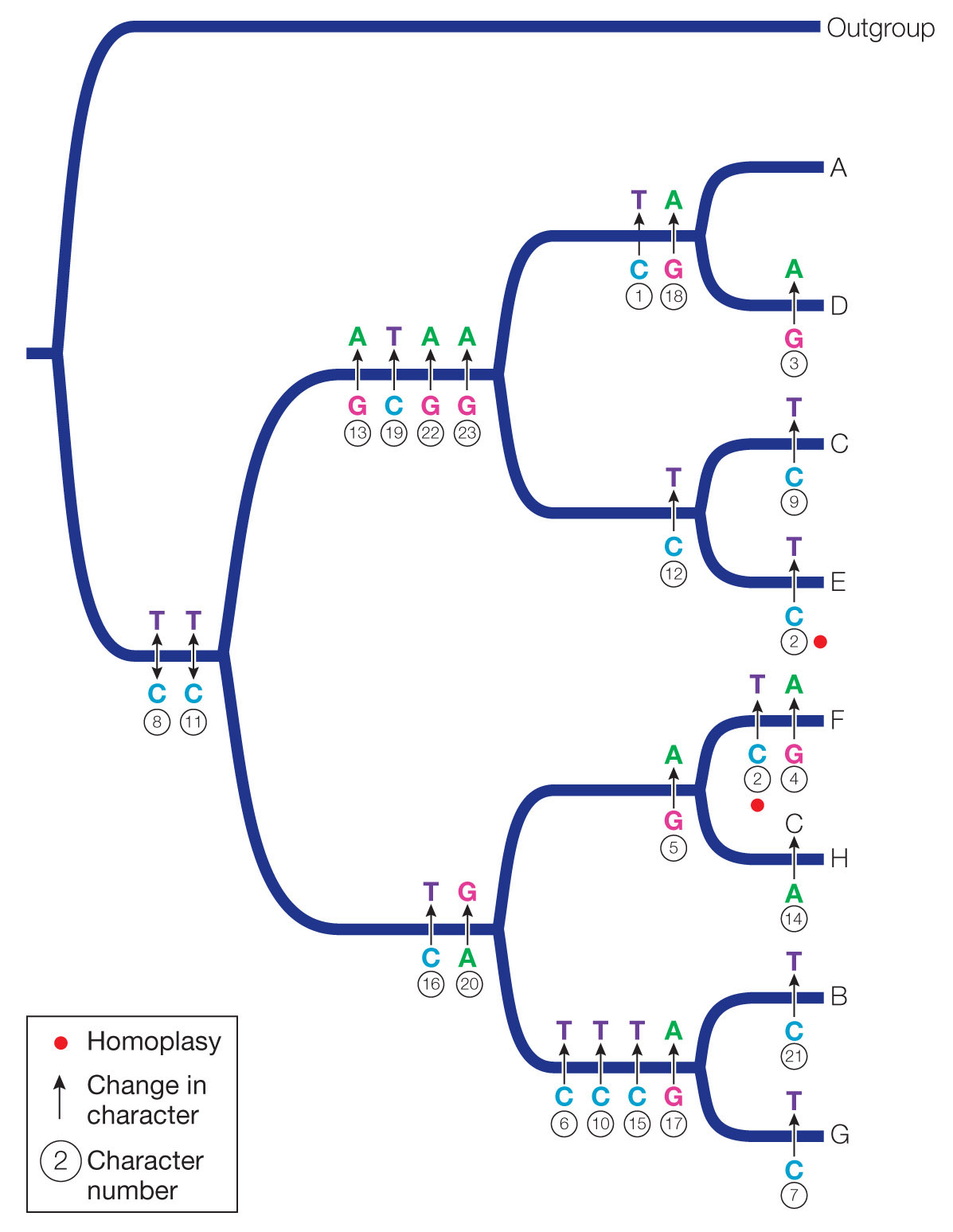
Ancestor of A and D: TCGGGCCCCCCCAACCGATACAA
Ancestor of C and E: CCGGGCCCCCCTAACCGGTACAA
Ancestor of F and H: CCGGACCCCCCCGACTGGCGCGG
Ancestor of B and G: CCGGGTCCCTCCGATTAGCGCGG
Ancestor of A, C, D, and E: CCGGGCCCCCCCAACCGGTACAA
Ancestor of B, F, G, and H: CCGGGCCCCCCCGACTGGCGCGG
Ancestor of A, B, C, D, E, F, G, and H: CCGGGCCCCCCCGACCGGCACGG
The reconstructed phylogeny requires 23 transitions and only 1 transversion. The viruses were grown in the presence of a mutagen, and the mutagen used in this experiment results predominantly in transitions.
FIGURE QUESTIONS
Figure 21.8 If the HIV sequences from the victim had been found to be more closely related to those of any other HIV-
Figure 21.12 The expected branch length would be about 0.09 substitutions per nucleotide.
APPLY WHAT YOU’VE LEARNED
The individual labeled CC01 is consistent with being the source of the infection, because that is the only individual with HIV sequences that are more closely related to HIV sequences in all of the other individuals, rather than to sequences sampled only within a single individual. Viruses from CC01 are found across the tree, which is consistent with CC01 transmitting viruses over time to the other individuals. (In the trial, CC01 was revealed to be the defendant in the case).
The viral sequences sampled from each of the other individuals in the cluster are monophyletic, indicating a single origin, with no further transmission to other individuals. Thus, the tree is inconsistent with CC02–
CC08 being the original source of infection in this cluster. The outgroup was needed to root the tree, which in this case identifies the direction of the transmission of events. Inclusion of viruses from outside the epidemiological cluster also tests the hypothesis that all viral sequences in this cluster are derived from a single ancestral virus.