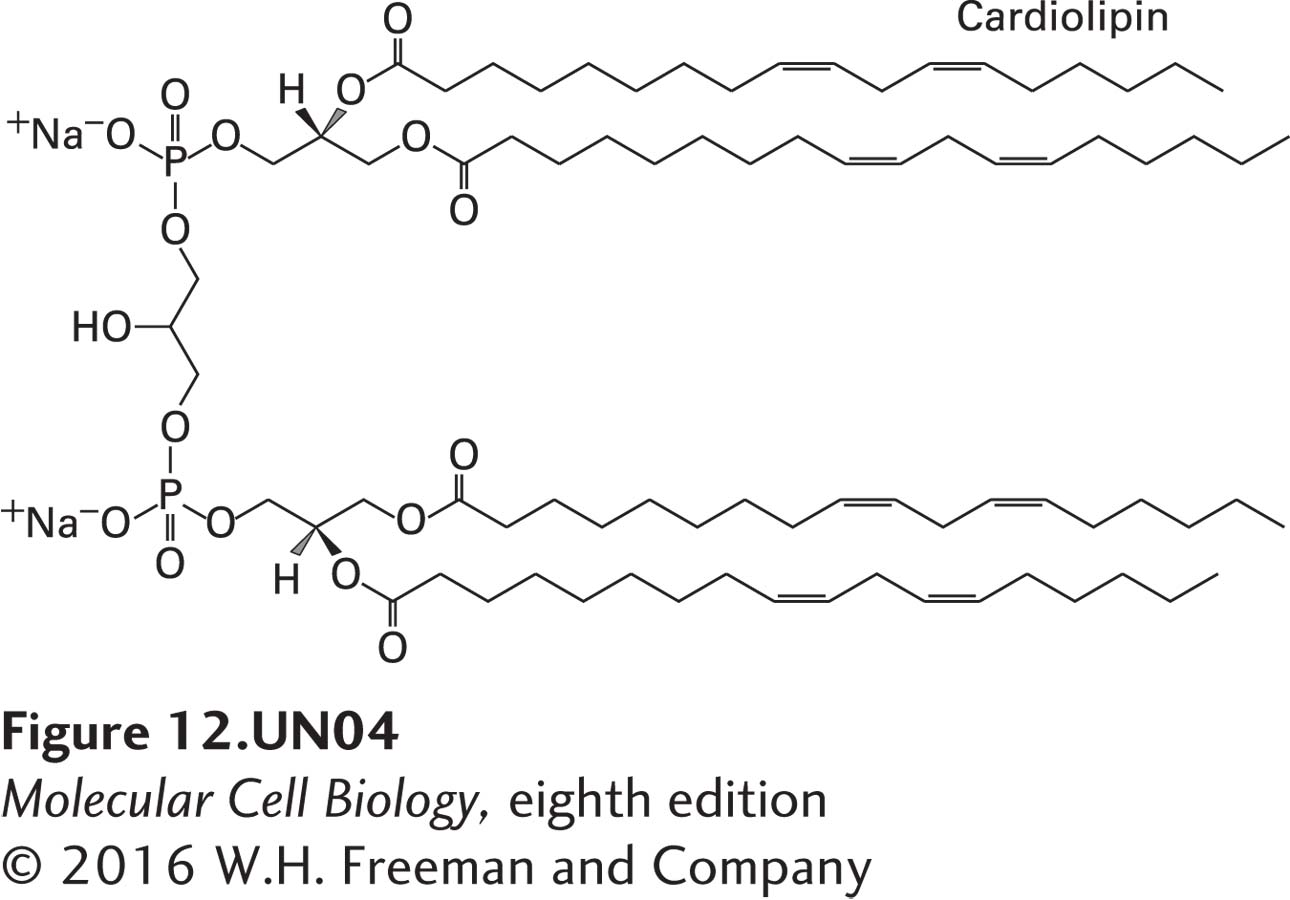
Over 50 years ago, Britton Chance proposed that the electron-transport complexes might assemble into large supercomplexes. Doing so would bring the complexes into close and highly organized proximity, which might improve the speed and efficiency of the overall electron-transport process. Indeed, genetic, biochemical, and biophysical studies have provided very strong evidence for the existence of electron-transport chain supercomplexes. These studies involved polyacrylamide gel electrophoretic methods called blue native (BN)-PAGE and colorless native (CN)-PAGE, which permit separation of very large macromolecular protein complexes, and electron microscopic analysis of their three-dimensional structures. One such supercomplex contains one copy of complex I, a dimer of complex III (III2), and one or more copies of complex IV (Figure 12-26). When this I/III2/IV supercomplex was isolated with ubiquinone (CoQ) and cytochrome c from BN-PAGE gels, it was shown to transfer electrons from NADH to O2; in other words, this supercomplex can respire—it is a respirasome. The precise function of supercomplex formation in the context of the very high protein concentration in the inner mitochondrial membrane remains to be established with certainty, but is thought to involve improving the speed and efficiency of electron transport, stabilizing individual multiprotein complexes, or preventing inappropriate protein aggregates.
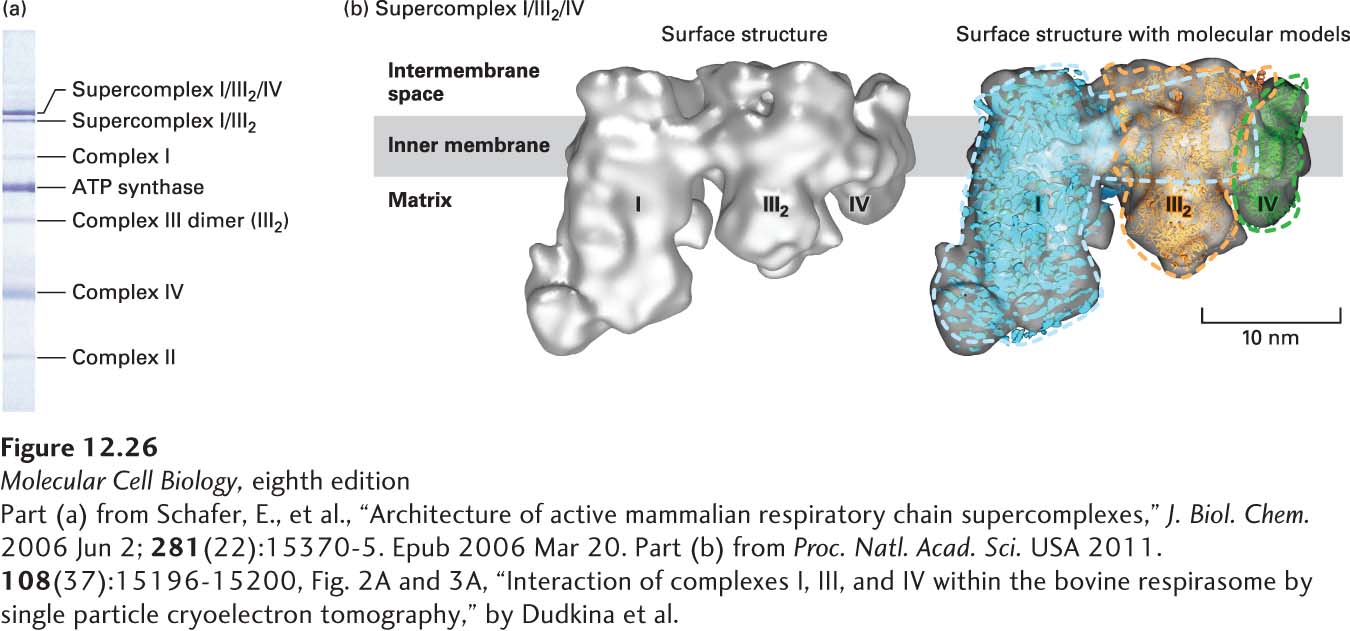
EXPERIMENTAL FIGURE 12-26 Electrophoresis and electron microscopic imaging have identified an electron-transport chain supercomplex containing complexes I, III, and IV. (a) Membrane proteins in isolated bovine heart mitochondria were solubilized with a detergent, and the complexes and supercomplexes were separated by gel electrophoresis using the blue native (BN)-PAGE method. Each blue-stained band within the gel represents the indicated protein complex or supercomplex. The intensity of the blue stain is approximately proportional to the amount of complex or supercomplex present. (b) Supercomplex I/III2/IV was extracted from a BN-PAGE gel, frozen, and visualized by cryoelectron tomography. The left image shows the three-dimensional surface structure viewed from an orientation parallel to the presumptive plane of the membrane. The right image is the same structure into which were fit models of the structures of the individual complexes: complex I (blue), dimer of complex III (III2, orange), and complex IV (green). Colored dashed lines represent the approximate outlines of these complexes. The complex I structure is based on essentially the entire complex I from the yeast Y. lipolytica, not just the 14 core subunits.
[Part (a) from Schafer, E., et al., “Architecture of active mammalian respiratory chain supercomplexes,” J. Biol. Chem. 2006 Jun 2; 281(22):15370-5. Epub 2006 Mar 20. Part (b) from Proc. Natl. Acad. Sci. USA 2011. 108(37):15196-15200, Fig. 2A and 3A, “Interaction of complexes I, III, and IV within the bovine respirasome by single particle cryoelectron tomography,” by Dudkina et al.]