The Topology of a Membrane Protein Can Often Be Deduced from Its Sequence
As we have seen, various topogenic sequences in integral membrane proteins synthesized on the ER govern the interaction of the nascent polypeptide chain with the translocon. When scientists begin to study a protein of unknown function, the identification of potential topogenic sequences within the corresponding gene sequence can provide important clues about the protein’s topological class and function. Suppose, for example, that the gene for a protein known to be required for a cell-to-cell signaling pathway contains nucleotide sequences that encode an apparent N-terminal signal sequence and an internal hydrophobic sequence. These findings suggest that the protein is a type I integral membrane protein and therefore may be a cell-surface receptor for an extracellular ligand. Furthermore, the implied type I topology suggests that the N-terminal segment that lies between the signal sequence and the internal hydrophobic sequence constitutes the extracellular domain, which probably has a part in ligand binding, whereas the C-terminal segment that lies after the internal hydrophobic sequence is probably cytosolic and may have a part in intracellular signaling.
Identification of topogenic sequences requires a way to scan sequence databases for segments that are sufficiently hydrophobic to be either signal sequences or transmembrane anchor sequences. Topogenic sequences can often be identified with the aid of computer programs that generate a hydropathy profile for the protein of interest. The first step is to assign a value known as the hydropathic index to each amino acid in the protein. By convention, hydrophobic amino acids are assigned positive values and hydrophilic amino acids negative values. Although different scales for the hydropathic index exist, all assign the most positive values to amino acids with side chains made up of mostly hydrocarbon residues (e.g., phenylalanine and methionine) and the most negative values to charged amino acids (e.g., arginine and aspartate). The second step is to identify long segments of sufficient overall hydrophobicity to be N-terminal signal sequences or internal stop-transfer anchor sequences and signal-anchor sequences. To accomplish this, the total hydropathic index for each segment of 20 consecutive amino acids is calculated along the entire length of the protein. Plots of these calculated values against position in the amino acid sequence yield a hydropathy profile.
Figure 13-16 shows the hydropathy profiles for three different membrane proteins. The prominent peaks in such plots identify probable topogenic sequences as well as their positions and approximate lengths. For example, the hydropathy profile of the human growth hormone receptor reveals the presence of both a hydrophobic signal sequence at the extreme N-terminus of the protein and an internal hydrophobic stop-transfer anchor sequence (Figure 13-16a). On the basis of this profile, we can deduce, correctly, that the HGH receptor is a type I integral membrane protein. The hydropathy profile of the asialoglycoprotein receptor, a cell-surface protein that mediates removal of abnormal extracellular glycoproteins, reveals a prominent internal hydrophobic signal-anchor sequence, but gives no indication of a hydrophobic N-terminal signal sequence (Figure 13-16b). Thus we can predict that the asialoglycoprotein receptor is a type II or type III membrane protein. The distribution of charged residues on either side of the signal-anchor sequence can often differentiate between these possibilities, since positively charged amino acids flanking a membrane-spanning segment are usually oriented toward the cytosolic face of the membrane. For instance, in the case of the asialoglycoprotein receptor, examination of the residues flanking the signal-anchor sequence reveals that the residues on the N-terminal side carry a net positive charge, thus correctly predicting that this is a type II protein.
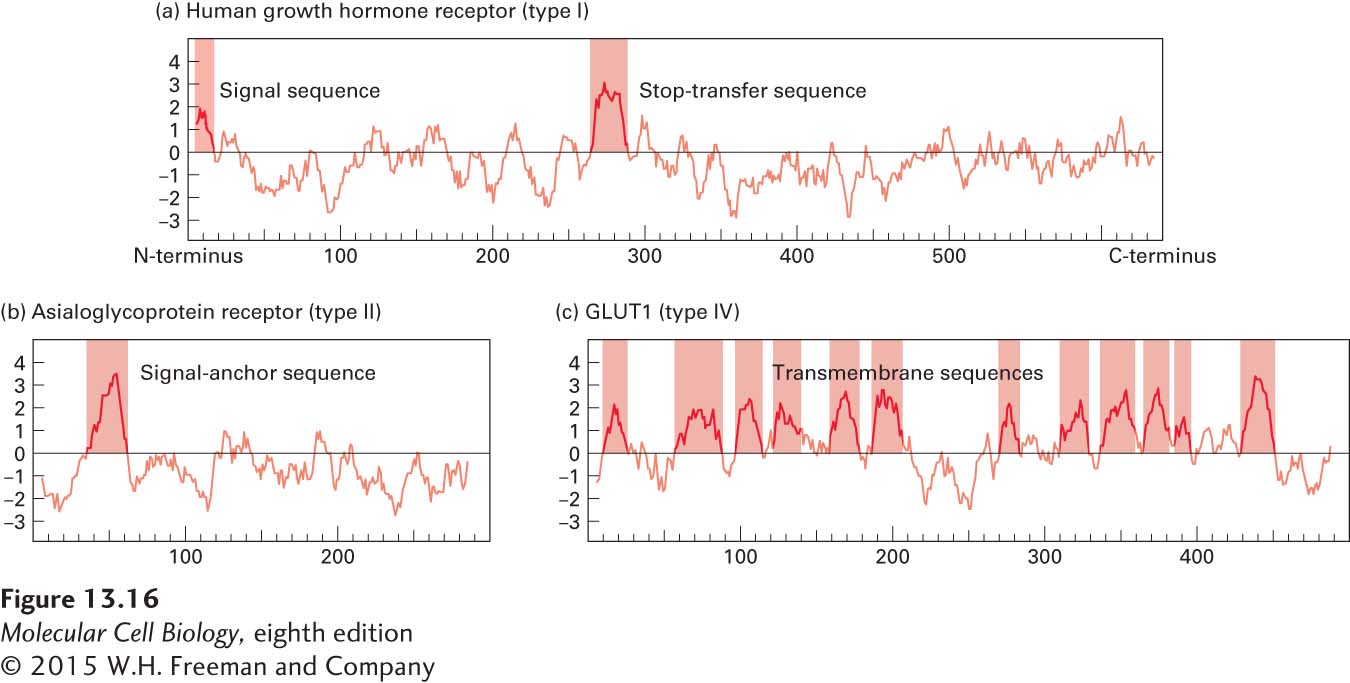
FIGURE 13-16 Hydropathy profiles. Hydropathy profiles can identify likely topogenic sequences in integral membrane proteins. They are generated by plotting the total hydrophobicity of each segment of 20 contiguous amino acids along the length of a protein. Positive values indicate relatively hydrophobic portions of the protein; negative values, relatively polar portions. Probable topogenic sequences are marked. The complex profiles for multipass (type IV) proteins, such as GLUT1 in part (c), must often be supplemented with other analyses to determine the topology of these proteins.
The hydropathy profile of the GLUT1 glucose transporter, a multipass transmembrane protein, shows the presence of many segments that are sufficiently hydrophobic to be membrane-spanning helices (Figure 13-16c). The complexity of this profile illustrates the difficulty both in unambiguously identifying all the membrane-spanning segments in a multipass protein and in predicting the topology of individual signal-anchor and stop-transfer anchor sequences. More sophisticated computer algorithms have been developed that take into account the presence of positively charged amino acids adjacent to hydrophobic segments as well as the length of and spacing between segments. Using all this information, the best algorithms can predict the complex topology of multipass proteins with an accuracy of greater than 75 percent.
Finally, sequence homology to a known protein may permit accurate prediction of the topology of a newly discovered multipass protein. For example, the genomes of multicellular organisms encode a very large number of multipass proteins with seven transmembrane α helices. The similarities between the sequences of these proteins strongly suggest that all have the same topology as the well-studied G protein–coupled receptors, which have the N-terminus oriented to the exoplasmic side and the C-terminus oriented to the cytosolic side of the membrane.