Unassembled or Misfolded Proteins in the ER Are Often Transported to the Cytosol for Degradation
Misfolded soluble secretory and membrane proteins, as well as the unassembled subunits of multimeric proteins, are often degraded within an hour or two after their synthesis in the rough ER. Initially, it was thought that proteolytic enzymes within the ER lumen catalyzed degradation of misfolded or unassembled polypeptides, but such proteases were never found. More recent studies have shown that misfolded secretory proteins are recognized by specific ER membrane proteins and are targeted for transport from the ER lumen into the cytosol by a process known as dislocation.
The dislocation and degradation of misfolded proteins depends on a set of proteins located in the ER membrane and in the cytosol that perform three basic functions. The first function is recognition of misfolded proteins. One mechanism for recognition involves the trimming of N-linked carbohydrate chains by mannosidases located in the ER (Figure 13-22). Trimmed glycans with the structure Man5–6(GlcNAc)2 are recognized by a protein known as OS-
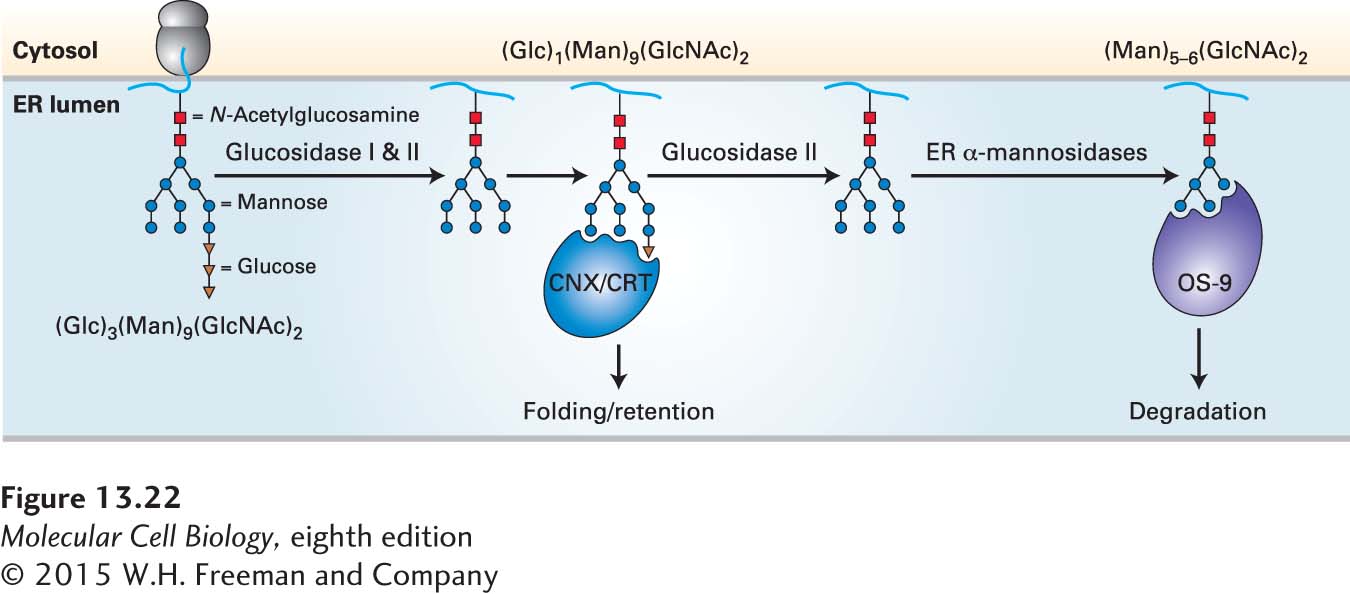
608
The second function required for dislocation of misfolded proteins is the transport of the marked proteins from the lumen of the ER across the ER membrane to the cytosol. A complex of at least four integral membrane proteins, known as the ERAD (ER-associated degradation) complex, enables dislocation of misfolded proteins across the ER membrane. The mechanism by which misfolded proteins traverse the ER membrane is not yet known, but there is no evidence that the ERAD complex forms a protein channel for dislocation, and it may be that dislocation involves powerful pulling and unfolding protein machines in the cytosol that drag misfolded proteins directly through the lipid bilayer.
Finally, as segments of a dislocated polypeptide are exposed to the cytosol, they encounter cytosolic enzymes that effect their degradation. One of these enzymes is p97, a member of a protein family, known as the AAA ATPase family, that couples the energy of ATP hydrolysis to disassembly of protein complexes. In dislocation of polypeptides from the ER, hydrolysis of ATP by p97 may provide the driving force to pull misfolded proteins from the ER membrane into the cytosol. As the misfolded proteins enter the cytosol, specific ubiquitin ligase enzymes that are components of the ERAD complex add ubiquitin residues to the dislocated peptides. Like the action of p97, the ubiquitinylation reaction is coupled to ATP hydrolysis; this release of energy may also contribute to trapping the proteins in the cytosol. The resulting polyubiquitinylated polypeptides, now fully in the cytosol, are removed from the cell altogether by degradation in proteasomes. The role of polyubiquitinylation in targeting proteins to proteasomes is discussed more fully in Chapter 3 (see Figure 3-31 and Figure 3-36).