Anchoring Proteins Localize Effects of cAMP to Specific Regions of the Cell
In many cell types, a rise in the cAMP level may produce a response that is required in one part of the cell but is unneeded, or perhaps deleterious, in another. Anchoring proteins localize members of the PKA family to specific subcellular locations, thereby restricting cAMP-dependent responses to these locations. Each of the roughly 50 such proteins, referred to as A kinase–associated proteins (AKAPs), has a two-domain structure; one domain confers a specific subcellular location and the other binds to the regulatory (R) subunit of PKA. AKAPs regulate cAMP and PKA signaling within the cell both spatially and temporally.
One AKAP in heart muscle anchors both PKA and PDE—the enzyme that hydrolyzes cAMP to AMP (see Figure 15-23)—to the outer nuclear membrane (Figure 15-31). Because of the close proximity of these two proteins, negative feedback provides tight local control of the cAMP concentration and hence of local PKA activity. As cAMP levels rise in response to hormone stimulation, PKA becomes activated and phosphorylates and activates several target proteins, including PDE. Active PDE, in turn, hydrolyzes cAMP, thus quickly returning PKA to its inactive state. The localization of PKA near the nuclear membrane also facilitates entry of its catalytic subunits into the nucleus, where they phosphorylate and activate the CREB transcription factor (see Figure 15-30).
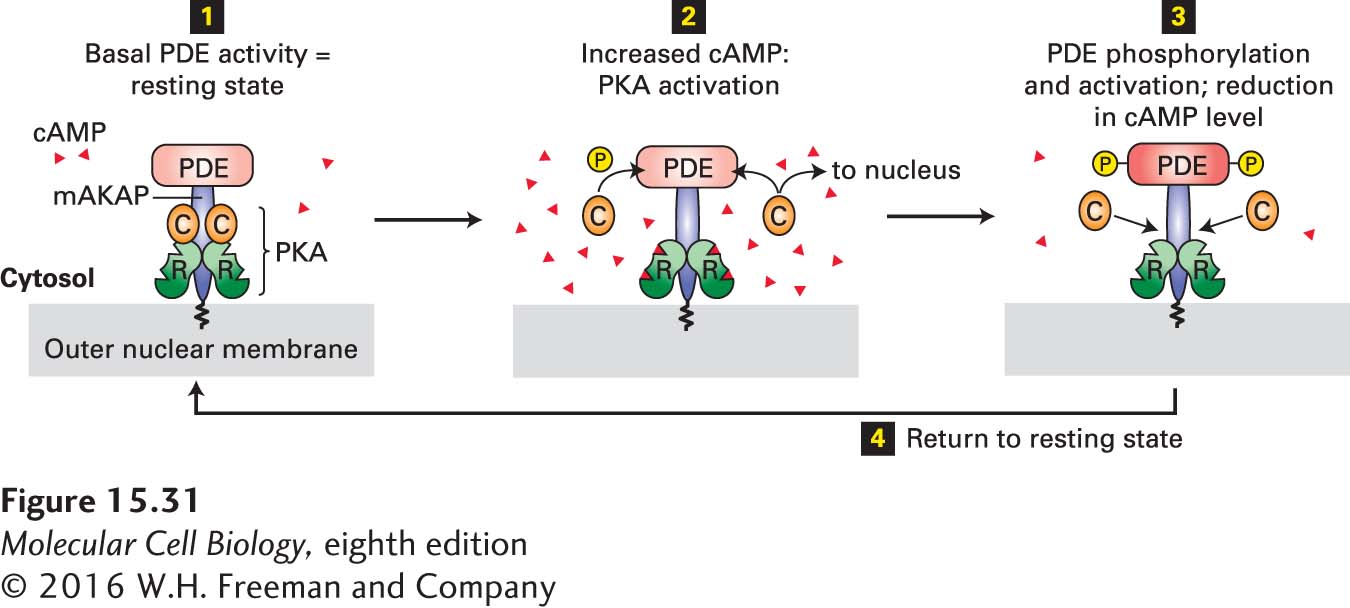
FIGURE 15-31 Localization of PKA and PDE to the nuclear membrane in heart muscle by an A kinase–associated protein (AKAP). This member of the AKAP family, designated mAKAP, anchors both PDE and the regulatory subunit (R, see Figure 15-27b) of PKA to the nuclear membrane, maintaining them in a negative feedback loop that provides close local control of the cAMP level and PKA activity. Step 1: The basal level of PDE activity in the absence of hormone (resting state) keeps cAMP levels below those necessary for PKA activation. Steps 2 and 3: Activation of β-adrenergic receptors causes an increase in cAMP to a level in excess of that which can be degraded by PDE. The resulting binding of cAMP to the R subunits of PKA releases the active catalytic (C) subunits into the cytosol. Some C subunits enter the nucleus, where they phosphorylate and thus activate certain transcription factors (see Figure 15-30). Other C subunits phosphorylate PDE, stimulating its catalytic activity. Active PDE hydrolyzes cAMP, thereby driving cAMP levels back to basal levels and causing re-formation of the inactive PKA C-R complex. Step 4: Subsequent de-phosphorylation of PDE returns the complex to the resting state. See K. L. Dodge et al., 2001, EMBO J. 20:1921.
In certain heart muscle cells, a different AKAP is tethered to the cytosolic face of the plasma membrane near a particular type of gated Ca2+ channel. In the heart, activation of β-adrenergic receptors by epinephrine (as part of the fight-or-flight response) leads to PKA-catalyzed phosphorylation of these Ca2+ channels, causing them to open; the resulting influx of Ca2+ increases the rate of heart muscle contraction. The binding of AKAP to PKA localizes the kinase next to these channels, thereby reducing the time that would otherwise be required for diffusion of PKA catalytic subunits from their sites of generation to their Ca2+-channel substrates.